E-learning 2. Analýza dat 2.2. Analýza vysokopokryvných genomických dat 2.2.1. DNA mikročipy 2.2.1.11. Analýza biologických drah
2.2.1.11.3 Metody analýzy genových sad a biologických drah
Nyní se budeme zabývat tím, jak lze rozpoznat, zda je daná genová sada (možná získaná z jednoho ze zdrojů zmiňovaných v předešlé kapitole) odlišně exprimovaná nebo ne. V posledních několika letech k tomuto problému již bylo navrženo velké množství metod, ale namísto jejich vyjmenování a procházení jedné po druhé, probereme raději hlavní charakteristiky, které tyto metody rozdělují.
2.2.1.11.3.1 Uzavřené vs kompetitivní metody
Vraťme se k naší mikročipové datové sadě Pima Indiánů. Jak jsme viděli v předchozí části, můžeme na tomto čipu identifikovat 96 sad sond, které odpovídají GO-termínu „inflammatory response“. Nejprve ze všeho co můžeme udělat, je kontrola výstupu statistického testu, který jsme již provedli. Například můžeme spočítat, kolik genů je významných na hladině významnosti 5%, tj. mají neupravené p-hodnoty pod 5%. Ukázalo se, že 8 z 96 genů je významných. Typickým způsobem převedení tohoto výsledku do p-hodnoty pro celkovou sadu genů je položení otázky: Jaká je pravděpodobnost pozorovat 8 významných genů z 96? Existují dva velmi odlišné způsoby, jak může být v této otázce pojem „náhoda“ interpretován. Těmito dvěma možnostmi je použít buď uzavřenou (self-contained) nebo kompetitivní (competitive) metodu, rozdíl a terminologii představili Goeman a Buhlmann [2007].
2.2.1.11.3.1.1 Uzavřené testování
U uzavřené metody používáme pouze hodnoty pozorované v rámci genové sady zájmu a ignorujeme všechny ostatní geny. Testovaná nulová hypotéza je
H0s = {”Žádný gen v genové sadě není odlišně exprimován”}
Pro geny, které nejsou odlišně exprimované je náhoda, že jsou významné na 5%ní hladině, takže můžeme očekávat, že 96 × 5% = 4,8 genů bude významných jen náhodou. Samozřejmě jsme pozorovali více než toto, ale je zde rozdílná významnost? K zodpovězení této otázky potřebujeme udělat předpoklad, že geny jsou stochasticky vzájemně nezávislé, tj. znalost hodnot pro jeden gen nám nedává žádnou informaci o jiných genech v genové sadě (je to docela silný předpoklad, který bude probírán detailněji níže). V tomto případě bude počet významných genů v sadě následovat B(96; 0,05) - rozdělení, tj. binomické rozdělení s n = 96 a p = 0,05. Pak můžeme podle tohoto rozdělení vypočítat p-hodnotu jako pravděpodobnost pozorování 8 nebo více významných genů a získáme p = 10,8%, tj. výsledek není na hladině 5% zcela významný (bylo by potřeba 10 z 96 genů).
2.2.1.11.3.1.2 Kompetitivní testování
V kompetitivním nastavení porovnáváme geny v rámci genové sady se všemi ostatními geny na čipu, tj. nulová hypotéza je
H0s = {”Genová sada není odlišně exprimovaná více než ostatní geny”}.
Porovnáním formulací těchto dvou nulových hypotéz je zřejmé, že jde v podstatě o zcela odlišné přístupy. Kompetitivní metoda například velmi závisí na velikosti mikročipu. Pokud máme velmi malý čip a na něm pouze 96 „inflammatory“ genů, není možné provádět kompetitivní test. Pokud je čip střední velikosti s tisícem genů, výsledek se bude lišit od celo-genomového čipu s 40000 geny atd. V našem případě můžeme provést kompetitivní test pozorováním, že z 12639 genů na čipu bylo celkem 1272 významných (10,1%), které signalizují, že je zde skutečně v genové expresi mezi dvěma skupinami značný rozdíl. Pokud náhodně vybereme 96 genů z čipu, můžeme očekávat, že 96×10,1% ≈ 9,7 genů bude významných jen náhodou, tj. v tomto případě je pozorovaný počet dokonce menší, než jsme očekávali podle nulové hypotézy. Data mohou být sumarizována do 2 × 2 kontingenční tabulky, uvedené v tabulce 2.2.1.11.2.
Tabulka 2.2.1.11.2: 2 × 2 kontingenční tabulka pro „inflammatory“ genovou sadu
.bmp)
U této tabulky testujeme, zda je nějaká asociace mezi jevem „Gen je významný“ a „Gen je v genové sadě“. Klasické testy řešící tento problém jsou Chí-kvadrát test nebo Fisherův exaktní test. Dřív se k výpočtu p-hodnot používal asymptotický výsledek, který není platný pro tabulky s malými očekávanými frekvencemi. Fisherův exaktní test používá exaktní hypergeometrické rozdělení, tj. pravděpodobnost pozorování nabízených marginálních součtů v dané tabulce. V našem případě je p-hodnota získaná z Fisherova testu p = 73,3 a tudíž daleko vyšší, než bylo pozorováno uzavřeným testem. I když se uzavřená hypotéza zdá být ze statistického pohledu mnohem přirozenějším způsobem formulování našeho testovaného problému, jsou kompetitivní metody široce užívané a s Fisherovým exaktním testem jsou možná dokonce celkově nejpopulárnějšími metodami.
2.2.1.11.3.2 Metody nezávislé na dělící hranici
Problémem metod popisovaných výše je, že jsou závislé na libovolném určení, které geny budou významné. Kdybychom například použili pro p-hodnotu 10%ní hranici, náš uzavřený binomický test by nám dal p-hodnotu 2,2%, tedy mnohem přesvědčivější výsledek. Přísnější hranice 1% by dala zase zcela jiný výsledek. Zkrátka se pouhým klasifikováním genů na významné nebo nevýznamné ztrácí množství potenciálně užitečné informace. Skutečnost, že 88 nevýznamných genů v genové sadě má p-hodnotu velmi blízkou 5% je velmi odlišná od situace, kde jsou tyto geny od významnosti daleko.
2.2.1.11.3.2.1 Uzavřený Kolmogorův-Smirnovův test
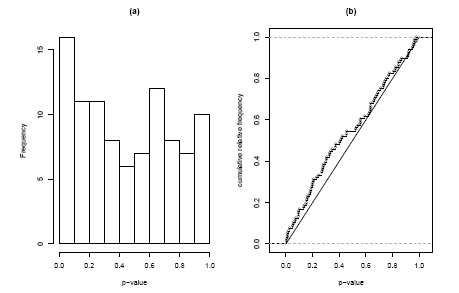
Řešení tohoto problému je použití úplné distribuce p-hodnot v našem testu. Na obrázku 2.2.1.11.4 (a) vidíme histogram 96 p-hodnot v „inflammation“ genové sadě. Podle uzavřené nulové hypotézy, že žádný gen v sadě není odlišně exprimován, bychom očekávali p-hodnoty, které by tvořili rovnoměrné rozdělení. Co místo toho vidíme, je náznak vrcholu na levé straně. K rozhodnutí, zda je tato odchylka od rovnoměrnosti významná, můžeme použít Kolmogorův-Smirnovův test. Tento test není založen na histogramu, ale na kumulativní distribuční funkci. Na obrázku 2.2.1.11.4 (b) vidíme pozorovanou kumulativní distribuční funkci v porovnání s očekávanou diagonální přímkou. Kolmogorova-Smirnovova statistika je dána maximálním rozdílem mezi těmito dvěma křivkami a odpovídající p-hodnota udává pravděpodobnost pozorování tohoto rozdílu, nebo víc, jestliže byly p-hodnoty skutečně rovnoměrně rozděleny. V našem případě získáme p = 8,2%.
Obrázek 2.2.1.11.4: Histogram a kumulativní distribuční funkce p-hodnot odpovídajících uzavřenému Kolmogorovu-Smirnovovu testu
2.2.1.11.3.2.2 Kompetitivní Kolmogorův-Smirnovův test
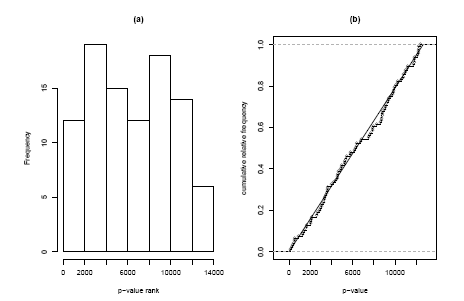
Nalézt kompetitivní protějšek je snadné nahrazením p-hodnot naší genové sady s jejich pořadím v rámci všech p-hodnot; tj. pokud má gen na čipu 835. nejmenší p-hodnotu, jeho pořadí bude 835. Podle kompetitivní nulové hypotézy jsou nyní tato pořadí předpokládána jako rovnoměrná přes čísla od 1 do 12639 (počet genů na čipu). Můžeme to znovu vizuálně zkontrolovat histogramem nebo kumulativním distribučním grafem (obrázek 2.2.1.11.5), ani jeden z nich neukazuje žádný zvláštní důkaz nerovnoměrnosti. Znovu použijeme Kolmogorovu-Smirnovovu statistiku k formálnímu testování a získáme p-hodnotu 85,1%, která náš vizuální dojem potvrdí. Dva velmi odlišné výsledky uzavřeného a kompetitivního testu se mohou zdát matoucí, ale musíme mít stále na mysli, že tyto testy zodpovídají dvě velmi odlišné otázky. Téměř významná p-hodnota uzavřeného testu říká „v této genové sadě může být nějaká odlišná exprese“, zatímco velmi nevýznamná p-hodnota kompetitivní analýzy říká „ale množství odlišné exprese není větší, než je na čipu obvyklé“. Tato tvrzení nejsou protichůdná v případě, kde se zdá být na čipu mnoho odlišně exprimovaných genů. Pokud tyto odlišné geny byly rovnoměrně rozděleny napříč dráhami přítomnými na čipu, můžeme v extrémním případě zjistit, že uzavřená metoda prohlašuje všechny dráhy za významné, zatímco kompetitivní metoda nenachází vůbec žádnou významnost.
Obrázek 2.2.1.11.5: Histogram a kumulativní distribuční funkce p-hodnot odpovídajících kompetitivnímu Kolmogorovu-Smirnovovu testu
2.2.1.11.3.3 Další problémy analýzy
Témata doposud probíraná mohou vysvětlovat nejzákladnější rozdíly mezi různými metodami analýzy genových sad, ale je zde počet dalších problémů, které je potřeba také zvážit. Nyní některé z nich rozebereme.
2.2.1.11.3.3.1 Směr testu
P-hodnoty z analýzy jednotlivých genů typicky pochází z dvoustranného testu, tj. rozhodneme se, zda je gen odlišně exprimován, ale neděláme rozdíl mezi zvýšenou nebo sníženou regulací. Takže když použijeme tyto p-hodnoty k vykonání analýzy genových sad, pak genová sada se stejným množstvím genů regulovaných směrem nahoru i dolů může být stejně významná jako jiná sada, která obsahuje pouze geny se zvýšenou regulací. Zda jsou změny v obou směrech v rámci genové sady důležité nebo ne, závisí na druhu genové sady a biologické situaci. Pokud nejsou, pak dříve zmíněná metoda může být lehce přizpůsobena k odhalení genových sad se zvýšenou nebo sníženou regulací za použití p-hodnot z odpovídajícího jednostranného testu.
3.3.2 Závislost mezi geny
Jak jsme již zmiňovali v části 2.2.1.11.3.1.1, doposud jsme předpokládali, že geny v rámci naší genové sady jsou podle nulové hypotézy stochasticky nezávislé. Což je docela nepravděpodobné, jelikož zvláště geny s podobnou funkcí nebo uvnitř stejné dráhy mají tendenci mít korelované profily genové exprese. Abychom viděli, jak to ovlivňuje metody, které jsme popisovali, zvažme extrémní případ, že všechny geny v genové sadě budou mít vždy identickou expresi. V tomto případě potřebujeme pouze analyzovat to, že jeden gen a jeho p-hodnota bude představovat kompletní sadu. Nicméně, jak binomický test, tak i Fisherův exaktní test budou mít buď p-hodnotu velmi blízkou nule (pokud p-hodnota genu je významná, a tudíž všechny p-hodnoty v sadě jsou významné) nebo blízkou 1 (pokud gen není významný), tj. tento test bude zcela zkreslovat skutečnost. Samozřejmě, že případ zcela korelovaných genů je nereálný, ale i mírnější korelace budou výsledky stále zkreslovat.
3.3.3 Převzorkovací metody
Většina přístupů k výpočtu p-hodnot nepoužívá asymptotickou nebo teoretickou distribuci, ale převzorkovací přístup. Za použití buď permutace čipů, nebo genů mohou být vypočítány nové verze datové sady, pro které je přepočítána statistika zájmu. Jsou vytvořeny všechny možné permutace nebo rozsáhlá náhodná sada těchto permutací, které nám udávají distribuci pro statistiku, která se dále používá k výpočtu p-hodnoty. Například Fisherův exaktní test je test, který je založen na převzorkování genů. Převzorkování genů je přímo spojeno s kompetitivní hypotézou, zatímco převzorkování čipů (vzorků) odpovídá uzavřené hypotéze. Problém korelace mezi geny může být řešen použitím převzorkování čipu. Pokud tento přístup použijeme k testování uzavřené hypotézy namísto binomického rozdělení, pak si převzorkované datové sady budou držet tuto korelační strukturu, a tudíž nám dají platnou p-hodnotu. Převzorkované geny v této situaci ovšem nepomohou, pokud nepředpokládáme, že jsou všechny geny na čipu vzájemně rovnoměrně korelované.
3.3.4 Vícenásobné testování
Dosud jsme popisovali analýzu pouze jedné genové sady (v našem příkladě šlo o sadu danou jedním GO-termínem). Avšak tento druh analýzy je obvykle prováděn současně na mnoha genových sadách, např. všechny GO-termíny, které náleží uzlu biologický proces nebo všechny lidské dráhy dostupné na KEGG. Při testování samostatných genů se musíme ujmout problému vícenásobného testování a to, že 1 ze 20 genových sadách bude určena významnou jen náhodou, pokud na p-hodnotu pro genovou sadu použijeme obvyklou 5%ní hranici. S analýzou genových sad se stavíme dalšímu problému: i když předpokládáme, že jsou samostatné geny stochasticky nezávislé, to stejné nebude pravda pro genové sady, které se často překrývají. Problém je zvláště závažný, když jde o GO-termíny, protože jsou hierarchicky uspořádány, tj. jistý termín definuje genové sady, které jsou podskupinami jiných sad. Tento problém je na detailní vysvětlení v tomto úvodním textu příliš složitý, ale měli bychom poznamenat, že jedna metoda může být v této situace použita bez jakéhokoli problému: obvyklá Bonferroniho metoda ke kontrole FWER (family wise error rate) nedělá žádný předpoklad o nezávislosti testů, takže pracuje za jakékoli situace genových sad, ačkoli může být velmi konzervativní.
3.3.5 Replikované geny
Gen je na většině mikročipů představován na více než jedné sondě, a často se počet kopií gen od genu liší. To samozřejmě může negativním způsobem ovlivnit analýzu např. pokud je jednotlivý gen ze sady odlišně exprimován, ale má více kopií na čipu, tak to způsobuje, že celá genová sada vypadá více změněná, než ve skutečnosti je. Nejlepším řešením tohoto problému je ještě před analýzou genových sad spojit hodnoty kopií do jednoho čísla. V případě Affymetrix čipů za použití Custom CDF anotačních souborů [Dai et al., 2005] pro částečné předzpracování se tento problém řeší, protože re-anotace čipů je navržena ke spojení všech sond představujících stejný gen. V jiných případech je široce užívaným řešením průměrování kopií. Povšimněte si, že takové průměrování by mělo být prováděno na původních normalizovaných hodnotách genové exprese a ne na souhrnných datech jako jsou p-hodnoty (průměr p-hodnot již nebude dále rovnoměrně rozdělen podle nulové hypotézy, tj. nebude to platná p-hodnota samo o sobě).