E-learning 2. Analýza dat 2.2. Analýza vysokopokryvných genomických dat 2.2.1. DNA mikročipy 2.2.1.7. Analýza expresních čipů
Důvod, proč hledat odlišně exprimované geny, byl zdůvodněn v předešlých kapitolách. Již víme, že se jedná o problém porovnávání skupin. Standardní metody k výpočtu velikosti účinku, testování hypotéz a regrese aplikované v porovnávání skupin byly definovány v kapitole 2.1.2. Porovnávání skupin. Připomínáme, že hlavní rozdíl mezi dvěma přístupy je, že testování hypotéz je analýza jedné proměnné, zatímco regresní metody jsou s více proměnnými. Často není exprese genů/proteinů ovlivněna pouze kategoriální proměnnou, ale také některými dalšími proměnnými. Jako jednoduchý příklad slouží účinek pohlaví nebo věku. Starší pacienti mají více aberací (odchylek) než mladší pacienti; geny na chromozomu X budou více exprimovány u žen v porovnání s muži. Proto, pokud víme o existenci takového faktoru, musíme s tímto efektem v analýze počítat. V tomto případě by měly být použity regresní metody.
Existují i další testování hypotéz a regresní metody, které byly navrženy speciálně pro expresní mikročipy. Tyto metody mohou být úspěšně použity i na všechny podobné problémy, jako například odhalování odlišné abundance proteinů mezi skupinami (data z hmotnostní spektrometrie, 2D gelové elektroforézy, atd...).
V následujícím textu popíšeme dvě další metody nebo porovnání skupin: SAM a LIMMA.
SAM (Significance Analysis of Microarrays)
SAM je v dnešní době široce užívaná metoda k určování změn v genové expresi. Je založena na upravení běžné, již dříve popsané t-statistiky.
Mějme matici hodnot genové exprese pro n vzorků a p genů: xij, i=1...p a j=1..n, kde sloupce představují vzorky a řádky geny, a skupinu proměnných yj, j=1..n, které rozdělují vzorky do dvou skupin, např. rakovinná a normální tkáň. Předpokládejme, že máme dvě skupiny, pak je upravená t-statistika použita v SAM definovaná jako
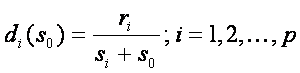
kde 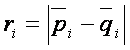 , pi a qi odpovídají průměrům genové exprese v každé skupině,
, pi a qi odpovídají průměrům genové exprese v každé skupině, 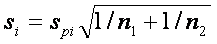 je obyčejná směrodatná odchylka pro obě skupiny a s0 je proměnný faktor, který musí být odhadnut. Tento faktor představuje rozdíl mezi t-statistikou a SAM. Z toho plyne, že pokud je s0=0, pak d je běžná t-statistika. Velmi velké hodnoty s0 dělají d ekvivalentní s t-statistikou bez čitatele. Cílem je odhadnout s0 tak, aby závislost d na si byla co možná nejmenší. Metodu k odhadnutí s0 lze najít např. v Tusher et al. 2001.
je obyčejná směrodatná odchylka pro obě skupiny a s0 je proměnný faktor, který musí být odhadnut. Tento faktor představuje rozdíl mezi t-statistikou a SAM. Z toho plyne, že pokud je s0=0, pak d je běžná t-statistika. Velmi velké hodnoty s0 dělají d ekvivalentní s t-statistikou bez čitatele. Cílem je odhadnout s0 tak, aby závislost d na si byla co možná nejmenší. Metodu k odhadnutí s0 lze najít např. v Tusher et al. 2001.
Když je SAM statistika vypočítána, musíme nastavit kritickou hodnotu pro významnost. Protože SAM statistika už není běžná t-statistika, nemůžeme použít běžný přístup kvantilů t-rozdělení. Proto je významnost hodnocena přes permutační postup navržený v Tusher et al. (2001).
Nejprve ze všeho jsou vzestupně řazeny SAM statistiky pro všechny geny: 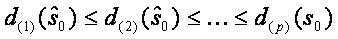 . Dalším krokem je permutace sloupců matice dat a přiřazení prvních n1 (počet vzorků ve skupině jedna) vzorků ke skupině 1 a zbytek n2 (počet vzorků ve druhé skupině) vzorků ke skupině 2. Celkem je provedeno n!=(n1+n2)! takových permutací. SAM statistika je pro každou permutaci popisována spolu s pořadím. Předpokládané uspořádaní statistiky podle nulové hypotézy, že není žádný rozdíl, může být odhadován jako
. Dalším krokem je permutace sloupců matice dat a přiřazení prvních n1 (počet vzorků ve skupině jedna) vzorků ke skupině 1 a zbytek n2 (počet vzorků ve druhé skupině) vzorků ke skupině 2. Celkem je provedeno n!=(n1+n2)! takových permutací. SAM statistika je pro každou permutaci popisována spolu s pořadím. Předpokládané uspořádaní statistiky podle nulové hypotézy, že není žádný rozdíl, může být odhadován jako
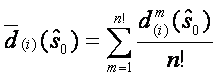
Každý gen i se svou SAM statistikou di(s0) značně větší než 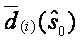 je považován jako odlišně exprimovaný. Označme tuto vzdálenost delta. Obvykle se to znázorňuje vykreslením očekávaných hodnot oproti pozorovaným hodnotám SAM:
je považován jako odlišně exprimovaný. Označme tuto vzdálenost delta. Obvykle se to znázorňuje vykreslením očekávaných hodnot oproti pozorovaným hodnotám SAM:
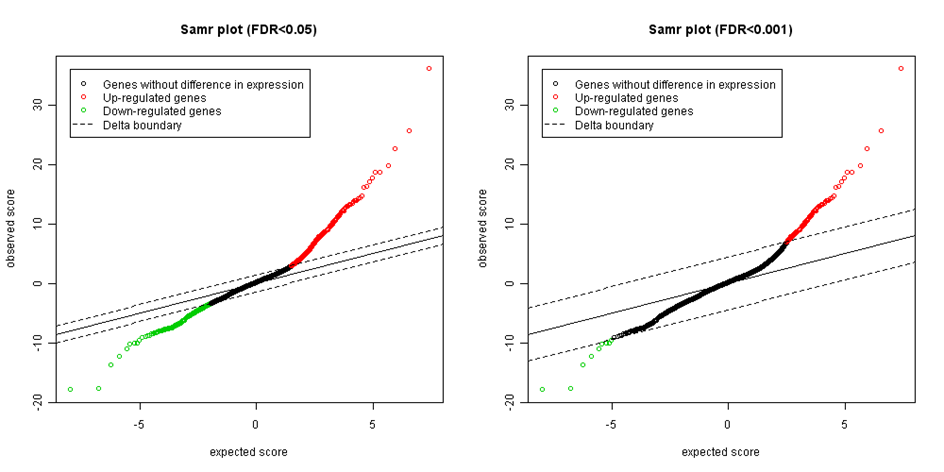
Obrázek: Očekávané versus pozorované SAM statistiky s různými hranicemi významnosti.
Konce na obou stranách představují možné významné geny. Čím více jsou body vzdáleny od přímky shodnosti (identity line) - černé diagonály, tím více jsou odlišně exprimovány. Posledním krokem je vhodné nastavení delta. Začneme s delta=0 a při současném kontrolování úrovně false discovery rate (FDR) tuto hranici postupně zvyšujeme. FDR je počítáno jako medián nebo 90. percentil počtu chybně zvolených genů dělených počtem genů významných. Existuje několik strategií, jak pro delta vybrat vhodnou hodnotu. Například nastavit medián FDR tak vysoko jak ji je vědec ochoten připustit (např. 5% nebo 1%), a pak vybrat nejmenší hodnotu delta odpovídající takovému FDR.
Po výběru delta se seznam významných genů vrací. Nicméně k filtrování genů s nebiologickým efektem by měla být vypočítána hodnota fold change.
Původní dokumentaci k SAM lze stáhnout zde:
Původní článek lze stáhnout zde:
Significance analysis of microarrays applied to the ionizing radiation response.
Pro více informací si prohlédněte webové stránky SAM:
http://www-stat.stanford.edu/~tibs/SAM/
Lineární modely pro mikročipovou analýzu (LIMMA)
LIMMA je balík metod pro analýzu odlišné exprese v mikročipech, založený na lineární regresi. Je vhodný na lineární model pro každý gen zvlášť a k poskytnutí stabilních výsledků v případě malé velikosti vzorku používá Empirické Bayesovské metody. Balík je implementován v R a poskytuje široký okruh funkcí jak pro normalizaci, tak pro analýzu odlišně exprimovaných dat. Lineární model a funkce odlišné exprese se aplikují na všechny typy mikročipů, i když jsou normalizační funkce dostupné jen pro dvoubarevné spotované mikročipy.
Použití limma k analýze odlišné genové exprese
Limma umožňuje vyjádření širokého okruhu lineárních modelů. Základní funkce vhodná na lineární model je lmFit. Funkce vyžaduje dva vstupy: matici dat s řádky představujícími profily genové exprese a sloupce představují vzorky a matici plánu (design matrix).
Matice plánu poskytuje znázornění modelu, který bychom chtěli přizpůsobit. Je založen na vysvětlujících proměnných, které mají být zahrnuty. Následují některé příklady, jak vhodně sestavit matici plánu. Má tolik řádků, jaký je počet vzorků v experimentu. Počet sloupců se liší v závislosti na návrhu modelu, např. zda používáme intercept nebo ne, kolik proměnných a jakého typu chceme v modelu zahrnout.
Matice plánu
Příklad jedné proměnné
Jestliže máme v plánu porovnat genovou expresi mezi dvěma skupinami, potřebujeme velmi jednoduchou matici plánu s pouze 2 sloupci. První sloupec bude obsahovat 1 ve všech řádcích, představující intercept; druhý sloupec bude obsahovat informaci o skupině, do které každý vzorek náleží. Intercept slouží jako referenční hodnota (základ), kterým porovnáváme všechny účinky, které chceme odhadnout. Vytvořme si pro názornost jednoduchý model. V pokoji je 50 mužů a 50 žen. Chceme vědět, zda jsou muži vyšší než ženy. Předpokládáme, že muži jsou v průměru vyšší než ženy, a sestavíme lineární regresní model založen na pohlaví. Model může být zapsán velmi jednoduše, jako
Výška =α + β*Pohlaví
Zde je α intercept, který představuje referenční hodnotu (v našem případě „průměrnou výšku žen“) a β je koeficient znázorňující, kolik centimetrů musíme přidat k průměrné výšce žen, pokud chceme získat průměrnou výšku mužů. Pohlaví je skupinová proměnná a představuje účinek mužů, tudíž má v případě žen hodnotu 0 (náš intercept představuje průměrnou výšku žen), v případě mužů je hodnota 1 (k získání průměrné výšky mužů, navýšíme průměrnou výšku žen o β).
V každém modelu je potřeba upřesnit, která skupina bude referenční, protože bude kódována v matici plánu jako 0. Druhá skupina bude kódována jako 1. Referenční skupina je obvykle ta, kterou porovnáváme, například normální tkáň, divoký typ bakteriálního druhu, nebo skupina pacientů, která odpovídá na určitou terapii. V našem příkladě jsme vybrali ženy. Matice plánu je v našem případě velikosti 100 x 2.
Intercept Skupina
1 1 0
2 1 0
... ... ...
49 1 0
50 1 0
51 1 1
52 1 1
... ... ...
99 1 1
100 1 1
Všimněte si, že první sloupec s čísly pořadí není součástí matice plánu.
Jestliže máme ve skupině proměnných více než dvě hodnoty, musíme v naší matici plánu upřesnit všechny možnosti, které mohou nastat. Například, když máme porovnávat genovou expresi mezi krevními vzorky tří skupin pacientů: zdraví (Z), pacienti s akutní myeloidní leukemií (AML) a pacienti s akutní lymfatickou leukemií (ALL). Protože chceme studovat rozdíly mezi všemi třemi skupinami, měla by být každá z těchto tří skupin zastoupena v modelu jednou proměnnou. Přirozeně jako referenci, kterou chceme porovnávat vybereme zdravé pacienty. Tyto hodnoty tedy budou zachyceny interceptem, takže můžeme souhlasit, že je to ve skupinové proměnné modelu znázorněno jako 0. Jak můžeme kódovat ALL a AML? Nemůžeme je kódovat 1 a 2, protože by to znamenalo, že skupina AML má dvakrát větší efekt na hodnoty exprese, než ALL. Můžeme kódovat pouze ano/ne, to znamená 0/1. K tomuto účelu vytvoříme tzv. „fuzzy proměnné“ ukládající všechny kombinace ve smyslu 0/1. V našem příkladě to znamená vytvořit dvě další proměnné, jednu kódující ALL (0-ne, 1-ano) a druhou kódující AML (0-ne, 1-ano).
Druhá je matice kontrastů, která umožňuje kombinovat do protikladů zájmu koeficienty definované maticí plánu.
Každý kontrast odpovídá porovnání zájmu mezi cílovými RNA. Pro jednoduchý experiment nemusí být matice kontrastů jednoznačně určena.
Zde si můžete přečíst původní dokumentaci o limma R balíku: