E-learning 2. Analýza dat 2.2. Analýza vysokopokryvných genomických dat 2.2.1. DNA mikročipy 2.2.1.10. Meta-analýza mikročipových experimetnů
Meta-analýza mikročipů je relativně nové téma. Vznikající metody jsou vytvářeny s konkrétním cílem. Podle našich znalostí neexistuje žádná studie, která by porovnala všechny dostupné metody. Cílem tohoto článku je poskytnout stručný přehled publikovaných metod a jejich SW implementací.
U každé metody byly sledovány následující vlastnosti: cíl a princip metody, typ mikročipových sklíček, maximální počet studií, které může metoda porovnat, vstupní data, výstupy a SW implementace.
S ohledem na výše zmíněné vlastnosti mohou být metody rozděleny takto:
Podle typu mikročipového experimentu:
- metody pro expresní nebo mikroRNA mikročipy
- metody pro CGH mikročipy
- metody pracující se seznamy názvů genů
- metody pracující s T-statistikami a p-hodnotami
- metody pracující s log2 podíly
- cDNA i Affymetrix mikročipy
- pouze Affymetrix mikročipy
- pouze cDNA mikročipy
- implementované v R
- implementované jako spustitelné soubory
- implementované ve WinBUGS
- 2
- 2 a více
- modelové přístupy
- klasifikátory
- skórovací metody
V následujícím textu rozdělíme metody podle typu mikročipového experimentu.
Metody pro expresní nebo mikroRNA mikročipy
Nejdůležitějším krokem při analýze expresních / mikroRNA mikročipů je nalezení odlišně exprimovaných genů, obvykle mezi dvěma nebo více skupinami vzorků. Tento krok zahrnuje obvykle testování hypotéz s úpravou na tzv. problém mnohonásobného testování. V důsledku tohoto testování získáme hodnoty testových statistik, p-hodnoty a seznam významně odlišně exprimovaných genů vybraných podle zvolené hladiny významnosti. Každý z těchto výstupů může sloužit jako vstup pro následující meta-analýzu. Proto jsme rozdělily metody expresních nebo mikroRNA mikročipů podle typu vstupních dat.
Metody pracující se seznamy názvů genů
Vstupní seznamy jsou seznamy významně odlišně exprimovaných genů nebo seznamy názvů všech genů seřazených podle hodnot testovacích statistik. Tyto metody jsou založeny na porovnání dvou nebo více seznamů genů nebo binárních vektorů.
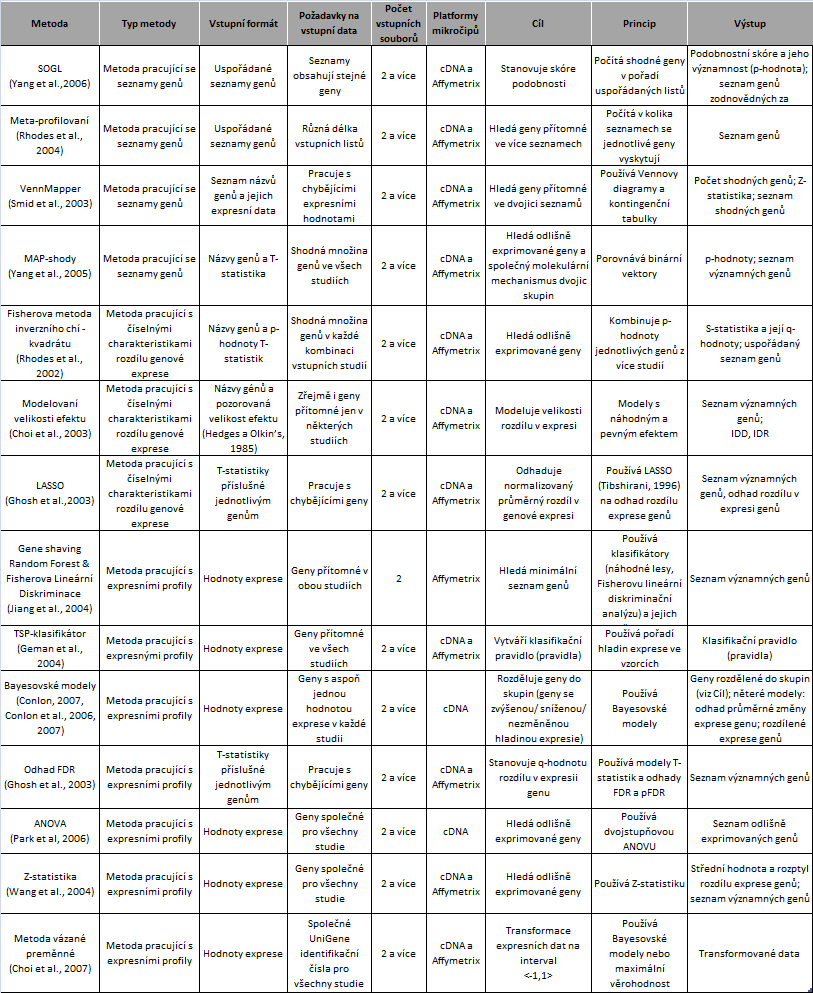
Jednou z prvních navržených metod je VennMapping metoda (Smid et al., 2003), která využívá Vennovy diagramy a kontingenční tabulky k nalezení genů přítomných ve dvojici seznamů odlišně exprimovaných genů. I když se vstup může skládat z více než dvou takovýchto seznamů, srovnání je provedeno vždy jen mezi dvojicemi seznamů. Metoda Rodese et al. (2004) označená jako Meta-profilování hledá geny přítomné v několika seznamech genů. Jedná se o modifikaci již dříve navržené metody Rhodose et al. (2002) (detaily jsou uvedeny v následující kapitole) používané pro vzájemné srovnání podobných mikročipových studií. Ve srovnání s tímto přístupem není Meta-profilování zaměřeno na ověřené analogové datové soubory, ale na srovnání a hodnocení průniku mnoha experimentů zkoumajících různé typy rakoviny - specifické datové soubory genových expresí. Cílem je najít rakovinné nespecifické geny zodpovědné za nádorovou transformaci. Princip je založen na výpočtení určité statistiky pro gen. Gen pronikne do algoritmu a tak může být zahrnut v konečném meta-seznamu významných genů pouze tehdy, pokud je významný v několika studiích. Nicméně, jak poukázal Yang et al. (2005), tato metoda je založená spíše na analýze významu genu, než na meta-analýze. Yang et al.,(2005) navrhl kombinaci VennMapping metody a Meta-profilování. Tato metoda se nazývá MAP-shody a k nalezení odlišně exprimovaných genů a běžných molekulárních mechanismů mezi odlišnými typy rakoviny používá analýzu binárních vektorů. SOGL (Yang et al., 2006) – počítá shody na první nebo poslední pozici seznamů genů, počítá skóre podobnosti pro seznamy genů.
Metody pracující s číselnými charakteristikami rozdílné exprese genů (hodnoty testových statistik, p-hodnoty)
Tyto metody jsou založeny na již známých konceptech meta-analýzy a modelování.
První navržená metoda byla Fisherova metoda inverzního chí-kvadrátu (Rhodes et al., 2002). Slučuje p-hodnoty – výsledky testování hypotézy - do S-statistik, které jsou poté použity pro testování hypotézy, že pozitivní výsledky z jednotlivých studií odpovídají stejným genům.
Další metodou používající modelový přístup: Modelování velikosti efektu (Choi et al., 2003) je metoda, která vytváří velikost efektu (Hedges and Olkin’s, 1985) modelem s náhodným nebo s pevným efektem a LASSO metoda (Ghosh et al., 2003) formující T-statistiky. Ta odhaduje parametry modelu pomocí LASSO metody (Tibshirani, 1996). Zpravidla je modelová statistika vhodná pro každý gen. Choi definuje IDD (Integration-Driven Discovery rate) a IDR (Integration-Driven Revison rate) stupeň. IDD je gen, který je identifikován jako odlišně exprimovaný pouze meta-analýzou (nebyl významný v žádné z předešlých analýz). IDR je podíl počtu IDD k počtu všech významných genů. IDD a IDR míry byly později používány k porovnání různých typů Bayesovských modelů. IDR byla definována Stevensem a Doergem (2005) jako gen, který byl označen významným v předešlých analýzách, ale nevýznamným v meta-analýze.
Tato skupina metod je velmi heterogenní a jednotlivé metody používají různé přístupy – od data-mining po běžné statistické přístupy, jako je ANOVA. Zde patří metody:
Některé z nich byly vytvořeny k nalezení genových markerů, které by mohly být použity ke klasifikaci nového vzorku například pomocí PCR. Tyto metody hledají minimální množství významných genů na rozdíl od jiných metod, které se zaměřují na hledání co největšího počtu významných genů. Mezi ně patří: metody GSRF (Gene Shaving Random Forests) a GSFLD (Gene Shaving Fisher’s Linear Discrimination) vytvořené Jiangem et al. (2004). Autor spojil tzv. Gene Shaving představenou Hastiem et al. (2000) a klasifikační metody. V GSRF a GSFLD se význam určitého genu stanoví podle poklesu míry chybné klasifikace tohoto genu.
Podobné cíle má TSP-klasifikátor (Geman et al., 2004), který vytváří klasifikační pravidla na základě exprese profilů. Klasifikační pravidla zahrnují páry genů a určují vztah mezi expresními hodnotami těchto dvou genů. Tato pravidla jsou poté použita v klasifikaci nových vzorků.
Colon ve svých pracích z roku 2006 a 2007 porovnává dva typy Bayesovských modelů (Conlon, 2007, Conlon et al., 2006, 2007). Vytvořil modely distribuce úrovně exprese každého genu. Geny mohl rozdělit na geny: se zvýšenou regulací, se sníženou regulací a shodně exprimované. Parametry Bayesovských modelů byly odhadnuty pomocí algoritmu Markovových řetězců Monte Carlo.
Ve stejném článku, který byl zmíněný u metody LASSO jsou také prezentovány metody založené na odhadu FDR (Ghosh et al., 2003). Tyto metody vznikly z modelů testových statistik a odhadů FDR a q-hodnot... Odlišně exprimované geny jsou určeny q-hodnotami.
Zatímco metody jako Choi et al.(2003) nebo Rhodes et al. (2004) nejsou velmi diskutovány a citovány, dvoustupňová ANOVA (Park et al., 2006) a Z-statistika (Wang et al., 2004) jsou... Dvoustupňová ANOVA nejprve odstraní variabilitu zapříčiněnou různými laboratořemi, a poté hledá odlišně exprimované geny testováním hypotéz (není žádný rozdíl v genové expresi mezi skupinami). Z-statistika odhaduje odchylku genové exprese smíšením expresních hodnot ze všech genů s podobnou průměrnou expresí, poté analyzuje rozdíl průměrné genové exprese mezi dvěma skupinami (použitím Z-statistiky). Myšlenka smíchání informací ze všech genů pochází z Bayesovského modelování.
Speciální metodou je metoda vázané proměnné (Choi et al., 2007). Jeden z problémů meta-analýzy mikročipových dat je rozmanitost rozsahu expresních hodnot pocházející z různých mikročipových platforem. Tento problém řeší právě metoda vázané proměnné. Přetransformuje data genové exprese do intervalu <-1,1> (pravděpodobnost exprese) metodou maximální věrohodnosti nebo Bayesovským modelováním. Transformovaná data jsou poté analyzována známými metodami analýzy mikročipů.
Přehled metod a jejich charakteristik ukazuje Tabulka č. 1.
Tabulka 1. Přehled metod meta-analýzy mikročipů
Metody pro meta-analýzu CGH mikročipů
Data pocházející z CGH mikročipů jsou více homogenní než data z expresních mikročipů. {Možná proto, že pro meta-analýzu CGH mikročipů neexistuje takové množství metod}. Je nám známá pouze jedna metoda meta-analýzy CGH mikročipů. Tato metoda transformuje data na jednotný formát a poté použije známé metody ke zjištění změněných segmentů CGH mikročipů. Transformace se skládá z těchto pěti kroků:
- Zmapování klonů
- Vyhlazení log2 podílů (Jong et al., 2004)
- Určení 100 pozic na každém chromozomu
- Přiřazení log2 podílů pozicím
- Konverze log2 podílů do Z-statistik