E-learning 2. Analýza dat 2.3. Analýza vysokohustotních proteomických dat 2.3.2. Hmotnostní spektrometrie 2.3.2.2. Statistické otázky v analýze proteomických profilů
2.3.2.2.2.1 Předzpracování
Před aplikací statistických modelů na MS profilová data musí být provedeno množství kroků, které analytická data předzpracují:
Kalibrace
Doba průletu (Time-of-flight) měřená spektrometrem je převedená na m/z měřítko pomocí množství kalibrů se známými m/z hodnotami.
Základní odčítání
Z matice je odstraněn základní „šum“, například lokálně aplikovanými vyhlazovacími technikami jako je Loess vyhlazování.
Normalizace
Aby byla vytvořena srovnatelnost mezi spektry, jsou data nějakým způsobem normalizována nebo standardizována. Cílem normalizace je odstranit technické zdroje variability (přístrojové chyby, odlišné množství vzorků, atd.). Velmi častý přístup je standardizování (lokálním nebo globálním) celkovým iontovým proudem. Kvůli možnému odstranění relevantní biologické informace, stejně tak jako požadované experimentální variability, se zájem o tento přístup zvýšil (Cairns et al, 2008). Optimální metody normalizace jsou předmětem pokračujícího výzkumu.
2.3.2.2.2.2 Zarovnání vrcholů a detekce
Jak na x (m/z) tak na y (množství) osách existuje nepřesnost, která kombinuje nebo srovnává profilové komplexy. Vrcholy v jakémkoli spektru jsou lokální maxima a mohou být definované jako body, které jsou maximálně +/- n bodů v okolí (budeme se na ně odkazovat níže jako na vrchol o velikosti n). V úvahu také musí být brán poměr signálu a šumu (SNR), to je obvykle stanoveno tím, že vrcholy musí určitým procentem překročit nějakou běžnou hranici hodnot intenzit.
I když mohou být vrcholy přijatelně nalezeny v každém individuálním spektru, jak mohou být vrcholy zarovnány napříč spektry umožňujícími variabilitu na ose x? Existuje mnoho metod detekce vrcholu, ale žádná z nich není perfektní. Například Morris et al. (2005) navrhl vytvoření samostatného spektra z průměru všech spekter a použití algoritmu detekce vrcholu založeného na vlnách tohoto samostatného složeného spektra. Tato metoda se vyhýbá problému zarovnání vrcholů a je založena na předpokladu, že pravý vrchol bude proti šumu v pozadí vyčnívat přes mnohonásobná spektra. Menší vychýlení mají za následek jen širší vrcholy. Tibshirani et al. (2004) navrhl aplikaci algoritmu jednorozměrného shlukování m/z hodnot vrcholů napříč spektry. Pevné shluky by měly reprezentovat stejný biologický vrchol, možná mírně posunutý na ose x. Střed každého shluku je v jejich metodě poté použit jako společná m/z hodnota pro tento vrchol.
V našem institutu jsme navrhli a aplikovali metodu, ve které je nejprve každé spektrum vyhlazeno použitím postupně se rozšiřujících klouzavých průměrů (Rogers et al., 2003; Barrett a Cairns, 2008). Vrcholy velikosti n v každém z těchto vyhlazených spekter (lokální maxima s nejméně n menšími body na každé straně) jsou identifikovány pro postupně větší hodnoty n. Rozložení četnosti je vytvořeno počtem detekovaných vrcholů v každém bodě, a podle tohoto konsenzu jsou určeny vrcholy. K ohodnocení vrcholů je pak každé spektrum porovnáno s výsledným seznamem vrcholů, aby se určila přítomnost/nepřítomnost tohoto vrcholu (uvnitř jistého okna tolerance). Je-li vrchol přítomen, pak je největší naměřená abundance uvnitř tohoto okna zaznamenána jako vrcholová abundance tohoto spektra. V každém spektru nemohou být všechny vrcholy detekovány. Předmětem zkoumání je, jak nakládat s „chybějícími“ vrcholy v následujících analýzách (například jako s nulami, chybějícími hodnotami nebo cenzurovanými pod jistou hranicí).
Po veškerém tomto zpracování jsou data z každého počátečního 2-D spektra redukována na vektor abundance mír pro pevně danou sadu obvykle několika stovek vrcholů.
Metody navrhnuté níže se vztahují k proteomickým studiím, kde je analýza založena na datech vrcholů a hlavními cíly jsou:
- Identifikovat vrcholy, které se liší mezi dvěma nebo více skupinami
- Vyvinout klasifikační schéma, které lze použít ke kategorizaci nových vzorků
2.3.2.2.2.3 Vícerozměrná ověřovací analýza dat
Data proteomických profilování jsou vícerozměrná, skládající se z měření početných inter-korelovaných vrcholů. Ověřovací analýza dat je vhodná k ověření velkých rozdílů mezi skupinami. Taková jednoduchá analýza slouží k vizuálnímu porovnání průměrů spekter dvou (nebo více) skupin.
Analýza hlavních komponent (PCA) může být v plné míře použita k prozkoumání vzoru variace. PCA je metoda pro snížení dimenzionality dat založena na rozložení kovarianční matice. První hlavní komponenta je lineární kombinace proměnných (v tomto případě vrcholových abundancí), která vyjadřuje největší podíl celkové variance; další komponenty jsou ortogonální lineární kombinace, které vysvětlují největší podíl zbylé variance.
To znázorňuje metoda použitá ke studiu transplantace ledvin (nepublikovaná data). Proteomické profily byly získány ze vzorků od 50 recipientů transplantovaných ledvin (30 s klinicky definovaným stabilním stavem a 20 s nestabilním stavem), 30 zdravých kontrol a 30 pacientů s ledvinovým selháním (před dialýzou), s podobným rozdělením věku a pohlaví v každé skupině a identickým protokolem pro zacházení se vzorkem a zpracování. Vzorky byly náhodně naneseny na čipy a všechny dvakrát analyzovány. Byly použity tři typy SELDI čipů (H50, IMAC a CM10). Vykreslením vzorků ve 2-D prostoru, který byl vytvořen prvními dvěma hlavními komponentami, můžeme vidět (na obrázku 2.3.2.4), jak dobře se skupiny separovaly.
Obrázek 2.3.2.4 Scatterplot prvních dvou hlavních komponent podle stavu pacientů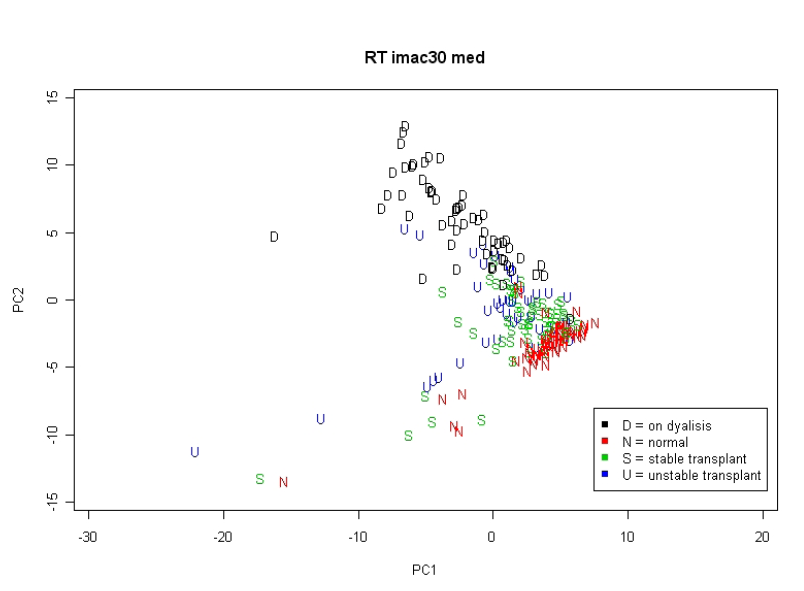
Je zřejmé, že se zdravé kontroly a pacienti se selháním ledvin ze separovaných shluků překrývají s pacienty po transplantaci, kteří se vyskytují uprostřed. Druhý graf, kde se analýza opakovala s použitím právě 50 pacientů s transplantací (obrázek 2.3.2.5) ukazuje, že stabilní a nestabilní pacienti nebyli dobře separováni.
Obrázek 2.3.2.5 Scatterplot prvních dvou hlavních komponent podle pacientů s transplantací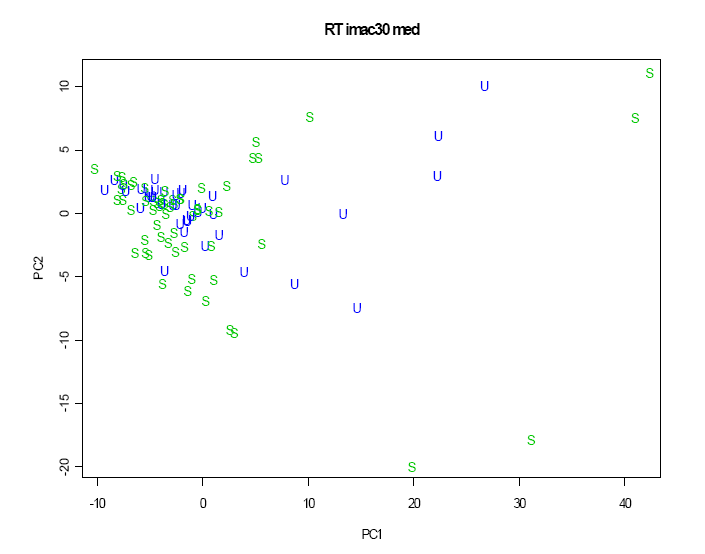
2.3.2.2.2.4 Statistické metody k detekování rozdílů mezi skupinami
Prvním cílem, kterému se zde budeme věnovat, je identifikování, které vrcholy (jestli vůbec nějaké) se liší mezi dvěma skupinami (např. případy a kontroly). Jednoduchým přístupem je provedení testu, jako je například dvouvýběrový t-test, k srovnání průměrů skupin pro každý vrchol. V případě, že bereme v úvahu duplikátní vzorky, může být rozšířen použitím modelu lineárních náhodných efektů.
Pro osobu i, replikaci k, je míra abundance yik modelována pro určitý vrchol jako
yik = α
kde xi je indikátor (1/0) skupiny (případ/kontrola) stavu, ηi je termín náhodného efektu odpovídající subjektu i a εik představuje reziduální varianci, včetně chyby měření. U posledních dvou pojmů se předpokládá normální rozdělení se střední hodnotou nula: ηi ~ N(0, σu2), εik ~ N(0, σe2). Test pro rozdíl mezi skupinami je proveden testováním, zda se β významně liší od nuly. Tento model poskytuje flexibilní rámec, ve kterém mohou být kovariáty jednoduše zahrnuty. Odhad intra-individuální variability je navíc proveden rozptylem σe2, který může být porovnán s celkovým rozptylem σe2 + σu2. Naopak odhad opakovatelnosti je proveden intra-skupinovým korelačním koeficientem σu2 / (σe2 + σu2), kde hodnoty blízké 1 signalizují shodu mezi duplikáty.
Výpočty velikosti vzorku
Velikost vzorku potřebná k dosažení určité síly k detekování odlišností mezi skupinami závisí na intra- (σ2) a inter- (τ2) varianci vzorku, počtu (m) technických replikátů a rozdílu ve středních hodnotách, které mají být detekovány (δ).
Pro porovnání dvou skupin, pro určitý vrchol, je počet vzorků n požadovaných v každé skupině
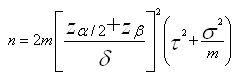
kde je α hladina významnosti a 1-β síla (Cairns et al, 2009).
K odhadu výpočtu velikosti vzorku jsou potřeba odhady variancí; požadavky velikosti vzorku závisí hlavně na intra- a inter- rozptylu vzorku mír abundance vrcholu.
σ2 a τ2 jsou odhadnuty použitím kontrolních dat. Nicméně existují vícenásobné (pravděpodobně několik set) vrcholy, které mají být zvažovány. Je použita ovšem pouze jedna velikost vzorku. Odhady rozptylu použité k výpočtu by mohly být mediánem, 90. percentilem nebo maximálním rozptylem napříč všemi vrcholy. Obecně je použití maxima příliš konzervativní a mediánu je nevystačující, protože vyjadřuje, že síla bude nižší než požadovaná pro 50% vrcholů. Dobrým kompromisem je proto 90. nebo 80. percentil. Odhad použitého rozptylu a také to, které percentily jsou ve výpočtu použity, má velký vliv na velikost vzorku potřebného pro studii. Další detaily a ilustrativní příklady výpočtů velikosti vzorku můžete najít v Cairns et al. (2009).
Vícenásobné testování
V proteomické profilové studii se typicky testuje několik set vrcholů. Ve stádiu objevování není cílem s jistotou demonstrovat rozdíl mezi skupinami, ale zajistit, že velká část „významných“ nebo zvýrazněných výsledků je skutečně pozitivní. Proto je vhodnější uvažovat o FDR (false discovery rate - očekávaném podílu falešných pozitiv mezi těmi, které jsou stanoveny za významné) než o FPR (false positive rate - očekávaném podílu falešně stanovených pozitiv mezi těmi, které jsou skutečně neplatné, odpovídající hladině významnosti).
Nechť P je celkový počet vrcholů a π podíl vrcholů, které jsou skutečně odlišně exprimované určitým množstvím, pak může být FDR odhadnuto jako:
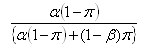
kde α je hladina významnosti a 1-β je síla. Tento vztah může být použit k volbě α za účelem dosažení určité FDR. Tabulky znázorňující vztah mezi FDR, α, β a π lze najít v Cairns et al. (2009).
Zlepšení statistického modelu
K postupu statistické analýzy byla navržena různá vylepšení (např. v Hill et al., 2008). Například normalizaci a případně i jiné kroky předzpracování lze provést ve stejném kroku jako testování a odhadování rozdílů mezi skupinami v jednotné analýze.
V experimentech, kde jsou známy identity proteinů a peptidů odpovídající vrcholům, může být struktura obsažena ve vrcholech (peptidy v rámci proteinů atd.)
2.3.2.2.2.5 Klasifikace vzorků
Existuje mnoho dostupných klasifikačních metod. My se zaměříme na Random Forest (RF) klasifikátor (Breiman, 2001).
V klasifikačním stromě jsou proměnné (v tomto kontextu míry abundance vrcholu) použity k postupnému rozdělení vzorků, dokud nejsou všechny subjekty ze stejného uzlu ve stejné třídě nebo dokud není dosaženo minimální velikosti uzlu. Vzhledem k velkému počtu proměnných porovnávaných s velikostí datového souboru bude obecně možné bezchybně klasifikovat vzorky, ale zásadní otázkou je vyvarovat se zašumělým datům.
RF algoritmus je klasifikátor založený na skupině klasifikačních stromů. V každém stromě jsou vzorky postupně rozdělovány použitím proměnné, která „nejlépe“ rozlišuje třídy. Obvykle je založen na Gini indexu, míře zanesení uzlů. Stromy mohou být vytvářeny, dokud se každý uzel skládá výhradně ze vzorků z jedné třídy, případně může být nejmenší velikost uzlu nastavena jako „stop pravidlo“. Idea RF je vytvořit mnoho stromů a klasifikovat na základě většinové volby. Aby se předešlo „over-fittingu“ a zavedení náhodných rozdílů mezi stromy v lese, jsou vytvořeny dvě modifikace. Zaprvé, je každý strom založen na „bootstrap“ vzorku původního datového souboru (tak, že z původního vzorku velikosti m, je bootstrap vzorek stejné velikosti získán vzorkováním s vracením). Zadruhé, je v každé části k výběru nejlepšího způsobu rozdělování dostupný pouze náhodný podsoubor proměnných. Každý strom je použit ke klasifikaci položek, které NEJSOU v bootstrap vzorku (tzv. out-of-bag položky); příznivou vlastností tohoto RF algoritmu je to, že poskytuje nezkreslený odhad klasifikačního výkonu.
Proteomická data rakoviny prsu
V roce 2007 se v Leidenu v Nizozemí setkalo několik skupin, aby porovnali metody klasifikace vzorků od pacientů s rakovinou prsu a kontrol na základě proteomických dat (Mertens, 2008). Vstupní datový soubor byl složený z MS proteomických profilů ze 76 případů s rakovinou prsu a 77 kontrol; profily z dalších 39 případů a 39 kontrol byly použity jako validační data. MS profily byly dostupné po předzpracování a skládaly se z mír abundance pro 11 205 bodů odpovídajících m/z podílům podél spektra.
Zde popisovaná analýza se sestávala z dvoustupňové strategie: detekce vrcholů, snížení 11 205 bodů na 365 vrcholů a následné aplikace RF algoritmu na data vrcholů. Pozornost byla zaměřena k minimalizaci závislosti strategie detekce vrcholů na vstupním datovém souboru. Podrobnosti metody jsou uvedeny v Barrett a Cairns (2008).
Použitím RF s 20 000 stromy v lese a klasifikováním podle většinové volby bylo správně zařazeno 84% vzorků. Důležité je, že když bylo použito stejné klasifikační pravidlo na nezávislou validační datovou sadu byla přesnost klasifikace velmi podobná (83% správně). Ve skutečnosti z metod použitých odlišnými skupinami, které se účastnily původního projektu, měla tato analýza (používající RF na vrcholy) nejnižší společné procento správných z původních dat, ale nejvyšší z validačních dat (Obrázek 2.3.2.6).
Obrázek 2.3.2.6 Klasifikace: kalibrační vs. validační výsledky
.PNG)
Ostatní použité metody zahrnují lineární a kvadratickou diskriminační analýzu, penalizaci logistické regrese a algoritmy podpůrných vektorů, z nichž se některé prováděly téměř stejně jako RF na validačních datech (ale nadhodnotily výkon na původním datovém souboru). Hodnocení metod lze nalézt v Hand (2008).