E-learning 2. Analýza dat 2.2. Analýza vysokopokryvných genomických dat 2.2.1. DNA mikročipy 2.2.1.8. Analýza CGH čipů
V poslední době se cílem studia mnoha biologických a lékařských experimentů stalo srovnání primární struktury dvou odlišných DNA - obecněji srovnání dvou vzorků DNA našeho zájmu. Takové porovnání může být provedeno ve srovnávacích studiích druhů/poddruhů (a použito v konstrukci fylogenetických stromů), protože se předpokládá, že genomové změny představují v evoluci druhů důležitou roli ([14], [5]). Ještě důležitější je, že porovnávání DNA je primárním nástrojem pro studia odhalující molekulární mechanismus některých vrozených nebo získaných onemocnění, zejména pak pro studia zabývající se složitostí nádorů.
Genetická nestabilita je jedním z klíčových faktorů v procesu kancerogeneze / maligní transformace buňky ([8], [34]). Zejména chromozomální aberace zahrnující delece, amplifikace, inverze a translokace jsou hlavní mechanismy aktivující onkogeny nebo inaktivující tumor supresorové geny, které jsou odpovědné za kontrolu buněčného cyklu, včetně proliferačních a reparačních procesů.
Rozsah ovlivnění genetické informace se pohybuje od jednoho nukleotidu (jednonukleotidový polymorfismus, SNP nebo jednonukleotidové změny) přes menší či větší chromozomální oblasti (změny počtu kopií) k celým chromozomům nebo chromozomovým sadám (aneuploidie).
Přítomnost těchto aberací úzce souvisí a může mít významný vliv na expresi genů ([17], [10]) a naopak [9].
Pro různé skupiny nádorů mohou být nalezeny specifické chromozomové aberace. Jedním dobře známým příkladem jsou mutace přítomné v BRCA1 nebo BRCA2 tumor supresorových genech, ty byly identifikovány jako důležité markery zainteresované v mnoha případech dědičných rakovin prsu a vaječníků. Ženy s určitými BRCA1 nebo BRCA2 mutacemi mají zvýšené riziko vzniku rakoviny prsu nebo rakoviny vaječníků [50]. Existuje řada takovýchto genetických markerů, z nichž některé mají všeobecný význam a lze je nalézt u mnoha typů rakovin, jiné jsou spíše tumor-specifické. Odhalení těchto změn je tedy důležité pro pochopení molekulárních mechanismů nemocí a pro nalezené konkrétních terapeutických cílů. Několik technik, jako jsou komparativní genomová hybridizace (CGH), „Representational Difference Analysis“ (RDA), fluorescenční hybridizace in situ (FISH) a nová technika CGH čipů nám umožňují tyto změny objevit. Technika CGH čipů je relativně nová a v dnešní době hojně používaná technologie pro identifikaci změn v genomu ([48], [38]). Hlavní výhodou této techniky ve srovnání s jinými metodami je, že umožňuje odhalit chromozomové aberace podél celého genomu v jednom experimentu.
2.2.1.8.1. Princip CGH čipů
Technika CGH čipů je zvláštním případem mikročipového experimentu.
Princip CGH čipů (viz obr. 1) vyplývá z tohoto schématu a je založen na komparativní genomové hybridizační (CGH) technice, která porovnává DNA z testovaných a referenčních vzorků (např. tumorových a zdravých tkání). Každá DNA je označena odlišným fluorescenčním barvivem, a poté hybridizována na čip s nanesenými imobilizovanými DNA sekvencemi zájmu. DNA z obou vzorků se kompetitivně hybridizují se sondami komplementárních sekvencí umístěných ve spotech. Mikročipová sklíčka jsou poté přečtena laserovým skenerem a použitím specifických filtrů jsou zachyceny fluorescenční intenzity obou kanálů a vytvořeny mikročipové obrazy. Tyto obrazy jsou poté analyzovány speciálním softwarem, který určí fluorescenční intenzity signálu z obou kanálů a převede je na číselná data. Jak již bylo zmíněno, fluorescenční intenzity každého spotu z obou vzorků jsou považovány za úměrné k množství přítomných genomových sekvencí vztahujících se k danému spotu. Pro další analýzu je vzat logaritmický podíl (log2 podíl) intenzit každého klonu z obou kanálů.
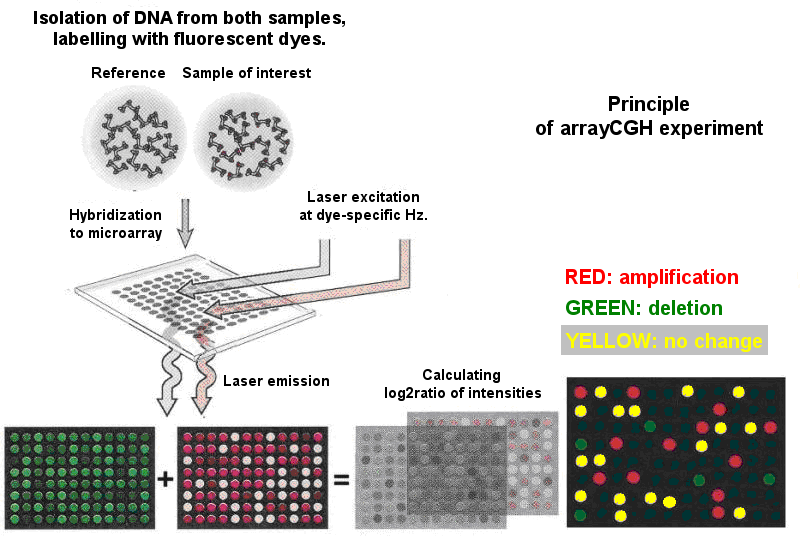
Obr. 1. Princip CGH čipového experimentu. Podle: http://www.mun.ca/biology/scarr/cDNA_microarray_Principle.jpg
{kind=link}
Technologie mikročipů může být použita nejen pro analýzu struktury, ale také pro aktivitu genomu. To lze provést jak na mRNA (expresních mikročipech) tak i na proteinové úrovni (proteinových mikročipech).
Existují různé technické i biologické příčiny variací, které mají na experiment vliv. Technické příčiny jsou způsobeny přípravou mikročipového sklíčka, stejně jako samotným procesem hybridizace, např. rozdíly účinnosti začlenění mezi různými barvivy, „prin-tip“ prostorovými efekty, intenzitou závislých účinků. Biologické rozdíly vyplývají z heterogenity buněk v kontrolním vzorku; nádorový vzorek je skoro vždy směs nádorových a normálních buněk. Heterogenita také existuje mezi nádorovými buňkami, protože různé části nádoru ukazují různé fáze progrese nádoru (tato heterogenita může mít za následek různé chromozomové aberace). Všechny tyto příčiny variability zkreslují data a zavádějí do nich šum. Předtím než mohou být data analyzována, musí být předzpracována, kvůli odstranění co největšího množství technických variabilit. To zahrnuje různé procesy filtrování s ohledem na kritéria kvality a normalizační požadavky.
2.2.1.8.2. Analyzování dat pocházejících z CGH čipových experimentů
Předpokládejme, že referenční vzorek pochází z normální buňky, a že v normální buňce existují dvě alely genu, log2 podíl roven nule znamená, že počet kopií genu testovaného a referenčního vzorku je stejný. Log2 podíl menší než -1 signalizuje deleci (log2 1/2=-1) a Log2 podíl větší než 0,58 amplifikaci genu v testovaném vzorku (Log2 3/2=0,58).
Jak již bylo zmíněno, hlavní předností CGH čipů je, že mohou odhalit chromozomové aberace podél celého genomu jen v jednom experimentu. Nicméně tato výhoda se může stát nevýhodou při současném analyzování všech výsledků. Nejjednodušší způsob, jak prozkoumat výsledky je vykreslit log2 podíly všech klonů v závislosti na příslušné pozici v genomu. Příklad takového zobrazení lze vidět na Obr. 2.
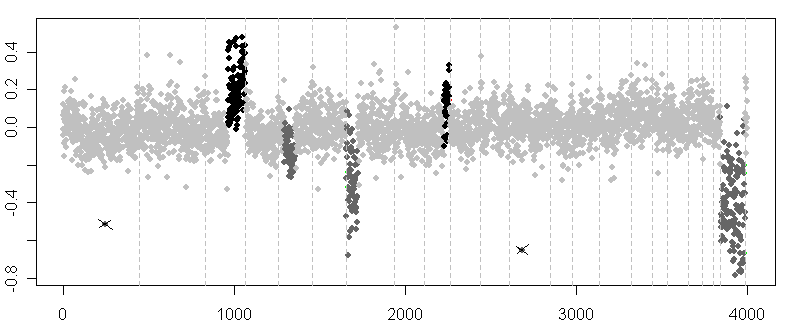
Obrázek 2. Graf log2 podílů klonů vykreslený v závislosti na pozicích v genomu. CGH čipový profil pacienta s Wilmsovým nádorem odhaluje několik deletovaných (tmavě šedé tečky) a amplifikovaných (černé tečky) oblastí s průměrným log2 podílem, který se zkresleně blíží k nule. Křížky jsou označeny dvě odlehlé hodnoty a přerušované svislé čáry představují hrany chromozomů.
Na obrázku je zobrazen CGH profil pacienta s Wilmsovým tumorem ([32], z veřejně dostupného datového souboru). Jak je vidět, teoretické hodnoty log2 podílů se zkresleně blíží k nule. To je způsobeno již výše zmíněnou technickou, a v tomto případě biologickou variací nádorového vzorku. Vzhledem k velkému počtu kontrolovaných genů a šumu v datech není jednoduchá vizuální kontrola grafu dostatečná k objektivní analýze aberací. Navíc, jak bylo uvedeno v [15], byl by takový přístup časově náročný, náchylný k selhání lidského faktoru a výsledky by nebylo možné opakovat.
Heurismus pro detekce deletovaných a amplifikovaných oblastí je použit k hraničním hodnotám odvozených z teoretických předpokladů o datech a jejich log2 podílech (v rozmetí od -1 do 0,58). Nicméně díky přítomnosti šumu se identifikace aberací stává při použití tohoto přístupu nesnadnou. Ke snížení dopadu šumu byly navrženy různé strategie (např. [38], [39]). Například prvně zmíněná odvozuje hranice z normálního vs. normálního čipu, vybráním genů s extremními log2 podíly. Hlavní nevýhoda prahovacích přístupů ovšem přetrvává; jednoduché umístění prahu pro detekci genových změn může vynechat klony ve změněných oblastech, ve kterých nejsou log2 podíly ani vysoké a ani dost nízké díky příliš vysoké variabilitě. Navíc, u některých z těchto přístupů je nutné provést jeden nebo více normálních/normálních experimentů.
Sofistikovanější metody pro objektivní analýzu CGH čipových dat jsou stále vyžadovány. Při hledání změn počtů kopií můžeme využít informaci o závislosti na pořadí genů v souvislosti s jejich skutečnými pozicemi na genomu. Vzhledem k tomu, že aberace obvykle ovlivní širší chromozomové oblasti, je rozumné předpokládat, že se u sousedních genů s největší pravděpodobností objeví stejná aberace. K tomu tématu bylo zveřejněno množství různých dokumentů používajících různých přístupů. Většina metod je založena pouze na převzetí závislosti na pořadí. Současný trend v těchto studiích je brát dodatečně v úvahu skutečné pozice genů v genomu (kvůli přesnějším výsledkům). Někteří autoři poukazují na to, že existují značné překryvy mezi klony v CGH čipovém experimentu a že se tyto překryvy dotýkají více než 30% velikosti klonu (Stejrnqvist et al., 2007). To by znamenalo, že předpoklad klonové nezávislosti je u většiny metod porušen. Další věcí zájmu jsou statistické významnosti zjištěných aberovaných oblastí, falešné odhalení výsledků (FDR) a klasifikace oblastí do jedné ze tří kategorií: deletované, amplifikované a bez aberace (později bude uváděna jako normální), nebo přesněji počtu změn kopií (delece jedné ze všech kopií, jednoduchá nebo násobná amplifikace kopie). U některých navržených metod je jedním z hlavních zájmů manipulace s odlehlými hodnotami. Zacházení s odlehlými hodnotami jako s hodnotami neodlehlými může významně ovlivnit zbývající údaje a celkový výsledek analýzy. Odlehlé hodnoty (viz obr. 1) mohou mít několik příčin. Odlehlé hodnoty mohou být výsledkem měření šumu nebo chybného označení klonů. Také je možné, že jedna odlehlá hodnota klonu odpovídá známé lokaci polymorfismu v počtu kopií (CNP). V neposlední řadě může odlehlá hodnota opravdu reprezentovat aberovanou oblast, to se stává ovšem zřídka. Metody pro analýzu CGH čipů jsou podrobně popsány v II. části. Obecně platí, že většina navržených metod předpokládá Gaussovo rozložení dat, pro které se může průměr (nebo dokonce i někdy směrodatná odchylka) změnit podle odlišné změny v počtu kopií.
Většina navržených metod je charakterizována čtyřmi hlavními koncepty: patří zde segmentační (také odhalení zlomových bodů) přístupy, přístupy využívající skryté Markovovy modely (HMM), přístupy využívající shlukování a vyhlazovací přístupy. Ačkoli mohou být metody klasifikovány podle jiných, výše zmíněných konceptů – jako je zavedení skutečné vzdálenosti, poskytující klasifikaci, atd. - budeme je členit podle dominantního přístupu.
V následující kapitole si detailně popíšeme hlavní přístupy analýzy CGH čipů. Předtím ale zavedeme některé pojmy používané v analýze CGH čipů.
2.2.1.8.3. Terminologie
Zatím jsme se setkali s následujícími pojmy: SNP (jednonukleotidový polymorfismus) pro změnu jednoho nukleotidu; amplifikace, delece nebo obecněji změny počtu kopií pro chromozomové aberace označující zisky nebo ztráty kopií v chromozomu; a aneuploidiemi označující změnu počtu kopií celého chromozomu. V literatuře se tato terminologie liší, obvykle v závislosti na cílu navrhované metody.
Někteří autoři rozlišují mezi snížením/zvýšením počtu kopií (podobně ztráty/zisky oblastí, amplifikace/delece), a používají tuto terminologii pro všechny typy změn počtu kopií, včetně aneuploidie. [3], [33], [53], [6], [21], [20], a [35] ] klasifikují změny počtu kopií v závislosti na počtu kopií přítomného genu: jedno a více nukleotidové delece, získáním jedné kopie a vyšší úrovení amplifikace, ale nakonec končí u třech tříd: delece, normál, amplifikace.
Někteří autoři jdou nad rámec tohoto rozdělení a zvažují také velikost aberací. Myers et al. [30] používá termín aneuploidie pro změnu počtu kopií celého chromozomu a segmentální aneuploidie pro dílčí chromozomální změny. Fridlyand et al. [15] rozlišuje čtyři hlavní typy aberací: : přechodné počty kopií, ztrátu a zisk celého chromozomu, fokální aberace a vyšší úroveň lokálních amplifikací. Také zdůrazňují, že každý typ je iniciován selháním různých molekulárních mechanizmů. U zisků nebo ztrát celých chromozomů se předpokládá výskyt selhání procesu dělení jádra nebo buňky. U mechanismu přechodu počtu kopií (která znamená nízkou úroveň a větší změny počtu kopií v rámci chromozomu), fokální aberace (nízká úroveň změn počtu kopií – menší než 1Mbp uvnitř chromozomu) a vysoké úrovně lokálních amplifikací (vysoké úrovně fokálních změn počtu kopií) je pravděpodobné, že byly zahájeny dvou-šroubovicovým zlomem DNA. Fridlyand se domnívá, že mezi zlomy vedoucích k fokálním aberacím a nízkými úrovněmi změn kopií existuje nějaký druh mechanického rozdílu.
Často používaná synonyma pro změny počtu kopií jsou také alternace počtu kopií, aberace počtu kopií a variace počtu kopií (CNV).
V následujícím textu se omezíme na termíny změny počtu kopií / delece / amplifikace a aneuploidie, pokud nebudeme popisovat konkrétní metody.