Special section Rare tumors Retinoblastoma
Definition
Retinoblastoma is a rare malignant congenital tumor of the embryonic neural retina, started by malignant transformation of primitive retina cells before final differentiation. It is the most common primary malignant intraocular tumor in childhood and predominantly affects infants and young children (Figure 1). Retinoblastoma is chemosensitive and radiosensitive tumor.
Retinoblastoma is responsible for 5 % of childhood blindness and 1 % of childhood mortality when it extends beyond the globe.
Retinoblastoma may occur as sporadic tumor or may be inherited.
Retinoblastoma is often curable when it is diagnosed early.
Patients with inherited retinoblastoma are at high risk to develop secondary cancer.
Epidemiology
Retinoblastoma accounts about 4% of all malignancies in children younger than 15 years
Incidence is about 1–4 : 1 million per year (1 : 18 000 – 30 000 live births)
No significant gender difference, M : F ratio is 1.12 : 1
Average age at diagnosis is 18 months, 90% of retinoblastomas are diagnosed in patients younger than 5 years
No race predilection appears for retinoblastoma
Mortality is reaching 50% worldwide, but there are significant differences between the countries depend on economic development of the country.
Etiology and etiopathogenesis
Retinoblastoma occurs in hereditary and nonhereditary form. The pathogenetic cause of retinoblastoma is the loss or mutation of the RB1 gene. RB1 gene is the tumor suppressor gene and plays a key role in cell cycle regulation, it is localised on chromosome 13q14.
- The functional loss of the RB1 gene caused by 13q14 deletion occurs in minority of patients with retinoblastoma
- The mutation of RB 1 gene is the most frequent cause of retinoblastoma. For normal function of the RB1 gene just one functional allele is enough, mutation in both alleles leads to malignant transformation of retinal cells (Knudson two hits hypothesis formulated in 1971) (Figure 2)
Nonhereditary (sporadic) retinoblastoma (about 2/3 of retinoblastomas) involve two acquired somatic mutations and it is not transmissible. It started at somatic level in a single retinal cell. Sporadic retinoblastoma characteristics:
- tumor is usually unilateral and unifocal
- median age at the time of diagnosis is 24 months
- patients with sporadic retinoblastoma have the RB1 mutation only on their retinal cells.
Hereditary retinoblastoma (about 1/3 of retinoblastomas) is caused by inherited mutation of germinal cells (germinal mutation) followed by the second acquired mutation of somatic cells. Patients with hereditary form of retinoblastoma have RB1 mutation in all their somatic cells. Hereditary retinoblastoma characteristics:
- 5–10% have positive family history of retinoblastoma (Figure 3)
- 90–95% have de novo (new) germinal mutation
- 85% are bilateral or multiple
- 15% are unilateral
- Very young age or in newborns (positive family - median age at diagnosis 4 months, for others hereditary retinoblastomas median age 13 months)
Hereditary form of retinoblastoma is transmitted as an autosomal dominant trait with 90 % penetrance, so almost 50% of children will be clinically affected.
For families with hereditary retinoblastoma genetic consultation and prenatal screening of RB1 mutation is recommended.
Association with other abnormalities or dysmorphism: the most children with retinoblastoma have no congenital abnormalities, but 5 % of children have retinoblastoma associated with other abnormalities:
- Syndrome associated with partial deletion of the long arm of chromosome 13 (syndrome 13q deletion) consist of microcephaly, hypertelorism, ptosis, micrognathia, broad nasofrontal bones – may leads to early diagnosis of retinoblastoma
- Mental retardation
- Failure to thrive
- Supernumerary fingers (polydactylia)
Clinical presentation and symptoms
The most common clinical signs and symptoms (more than 90 % of retinoblastomas) are:
- Leukocoria 60% (white pupillary reflex or „cat’s eye reflex“) usually seen on photograph – (Figure 4)
- Squint 20% (strabismus, secondary to macular involvement), usually convergent, at times divergent
- Glaucoma (secondary increased intraorbital pressure), cataracta
- Bulging of the eye, proptosis
- Loss of vision
- Different color in each iris
- About 10 % of children develop symptoms similar to orbitocellulitis or other infections (toxocarosis) – so called „pseudoretinoblastoma“:
- Redness of the eye (secondary to intraocular inflammation or vitreous hemorrhage)
- Persistent eye pain
Unusual clinical presentation include trilateral retinoblastoma consist of solitary midline intracranial (usually pineal) retinoblastoma. It is associated with hereditary retinoblastoma, and there is a latent period between diagnosis of intraocular retinoblastoma and trilateral retinoblastoma. Patients with trilateral retinoblastoma usually develop intracranial metastases and prognosis is very poor.
Diagnostic procedures
Initial studies for retinoblastoma should give the answer to question:
- extent of intraocular tumor
- locoregional extrabulbar involvement (presence or absence of orbital extension, optic nerve involvement )
- dissemination of disease (CNS, lungs, regional lymph nodes, liver, bones, bone marrow)
Personal and family history: information regarding family history are very important for further diagnostic decision and management. Family history is mandatory for genetic testing not only the patient, but also other family members and siblings.
When retinoblastoma is suspected, patient has to be sent to specialized pediatric oncology center for further specialized investigations.
Ophthalmology examination: by indirect ophthalmoscopy (in general anesthesia) is important to evaluate both eyes, to measure tonometry to exclude secondary glaucoma. Ophthalmoscopy in details describe localization and size of retinoblastoma and can detect intraocular extent of disease (vitreal seeding) (Figure 5) Ophthalmoscopic findigs are recommended to document with photography.
Ocular ultrasound is helpful in detection of intratumoral calcifications
Laboratory tests: there are no specific tumor markers or other specific laboratory test for retinoblastoma. Usual blood tests and biochemical tests are routine part of work up.
Imaging studies::
- CT scan : demonstrate a solid intraocular pathology mass usually with characteristic calcifications affecting retina and may detect involvement of intraocular structures (vitreous gel)
- MRI scan can detect mild hypointense mass in T1 weighted images, strong hypointense mass in T2 weighted images, but is unable to detect calcifications. MRI is important for detection of optic nerve involvement (Figure 6)
- Assessment of extraocular extent of disease: usually bone scan and bone marrow aspiration and biopsy and lumbar puncture with CSF (cerebrospinal fluid) examination is a part of initial work up for retinoblastoma
Histopathology of the tumor and molecular genetic for RB1 gene mutation is essential for correct diagnosis.
Differential diagnosis
- retinopathy of premature children (Figure 7)
- uveitis
- toxocariasis
- vitreoretinal dysplasia
- other tumors (retinal astrocytoma, optic glioma, retrobulbar neuroblastoma or soft tissue sarcoma)
Therapy
The main goals of therapy for retinoblastoma:
- save the life
- preserve vision or salvage eye, to ovoid enucleation („eye-free survival“)
- to minimize any complications and late side effects
Treatment options
- enucleation/ orbit exenteration
-
local intraocular therapy:
- cryotherapy/thermotherapy, laser and photocoagulation, plaque radiotherapy
- external radiotherapy
-
chemotherapy:
- sub-tenon application of carboplatin intraarterial application of chemotherapy (retinal arthery)
- systemic chemotherapy
Surgery: radical resection of the tumor for retinoblastoma means enucleation. Bigger impact on whether the affected eye (and vision in the eye) can be saved has the stage of the tumor according to International classification for intraocular retinoblastoma (ICRB) . It divided intraocular retinoblastoma in to 5 groups (A – E).
Enucleation is indicated in case of:
- active vital tumor in blind eye
- massive intracular involvement (vitreous seeding) (Figure 8)
- secondary glaucoma caused by tumor invasion
- as a salvage option when other therapy is ineffective
Local intraocular therapy: is in the hands of ophthalmologists and depends on size, number and localization of retinoblastoma (Figure 9)
Chemotherapy: chemoreduction is effective for intraocular tumors hand in hand with local intraocular therapy. For locoregionally advanced and disseminated retinoblastoma is chemotherapy the treatment of choice.
External radiotherapy the role of external radiotherapy was significantly reduced due to potentiation of risk of secondary malignancy (osteosarcoma). External radiotherapy is no longer a common treatment for retinoblastoma. It could be indicated in case of:
- monolateral sporadic small tumors localized in macula region (in order to safe the vision)
- salvage therapy and ineffective other therapeutic options
- palliative setting
Prognosis and outcome
Most of the retinoblastomas are curative tumors, 5-year overall survival rates for retinoblastoma range 86–95%.
Survival depends on :
- Hereditary or sporadic retinoblastoma
- Bilateral or monolateral disease
- Extent of intraocular disease or loco-regional disease
- Metastatic disease (extraocular extension is the most important risk factor for death).
5-year overall survival:
- 85–95% for localized intraocular tumor
- 60% for optic nerve involvement
- 20% for positive surgical margins
Patients with trilateral retinoblastoma and CNS involvement have median of survival only 8 months.
Patients with hereditary retinoblastoma and secondary tumor development (most common osteosarcoma) have 5-year overall survival less than 50%.
Author: Viera Bajčiová, MD, PhD
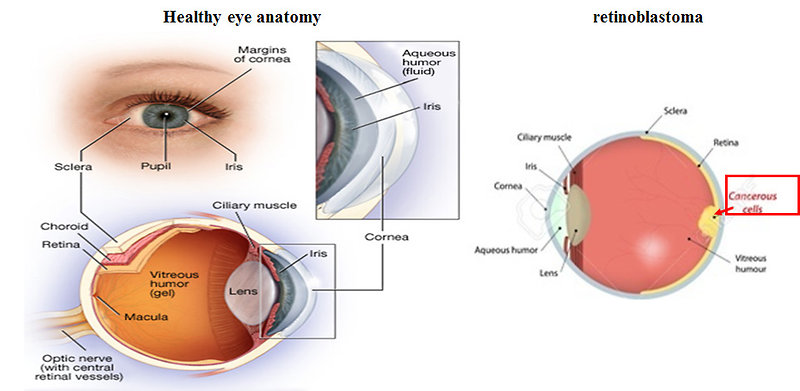 Figure 1: Anatomy of the eye and retinoblastoma (www.medscape.com) |
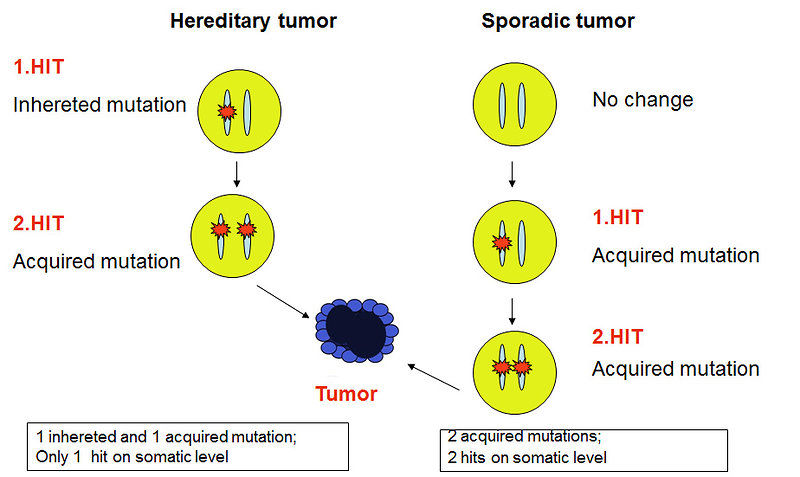 Figure 2: Knudson two hits hypothesis (Bajčiová, genetic syndromes 2015) |
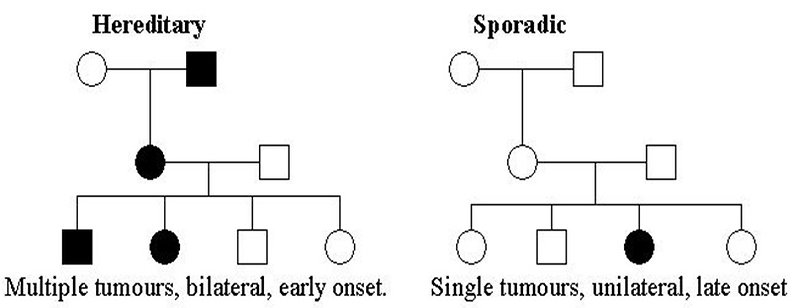 Figure 3: Pedigree of sporadic and hereditary retinoblastoma (KDO FN Brno) |
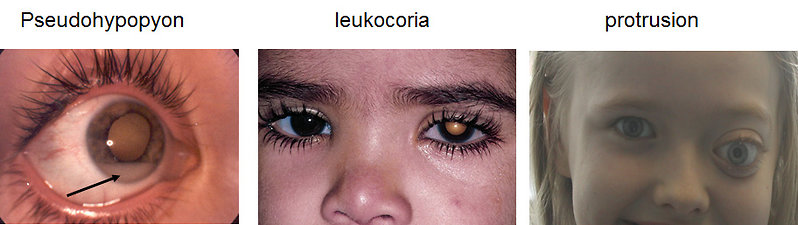 Figure 4: Clinical presentation of retinoblastoma (KDO FN Brno) |
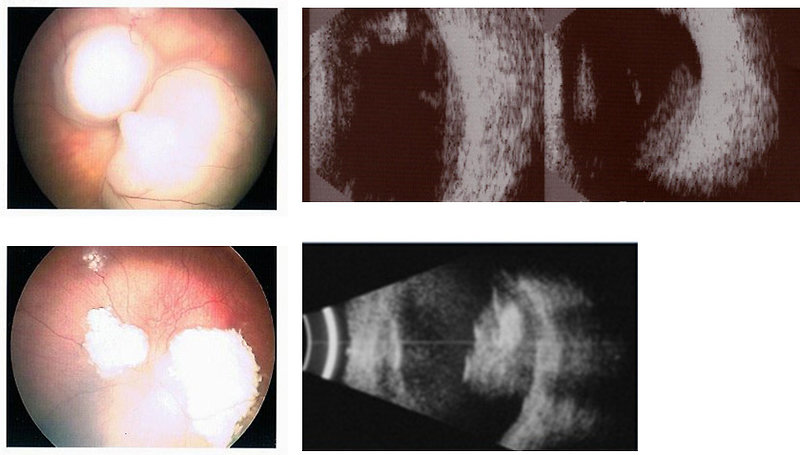 Figure 5: Ophthalmoscopy and ocular ultrasound (Morley J, www.wikimedia.org, 2008) |
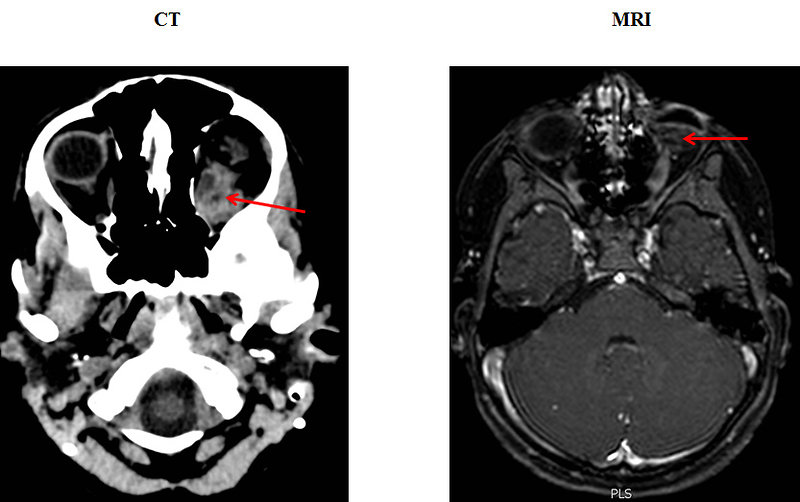 Figure 6: CT and MRI of retinoblastoma (KDO FN Brno) |
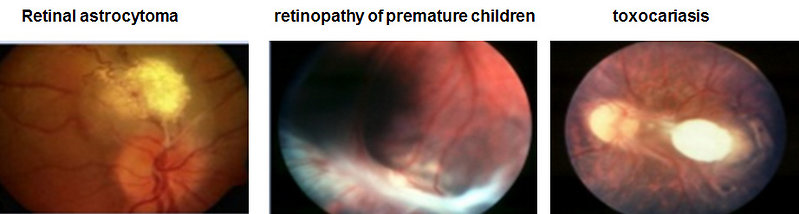 Figure 7: Differential diagnosis of retinoblastoma (Nurul I, Health and medicine 2014) |
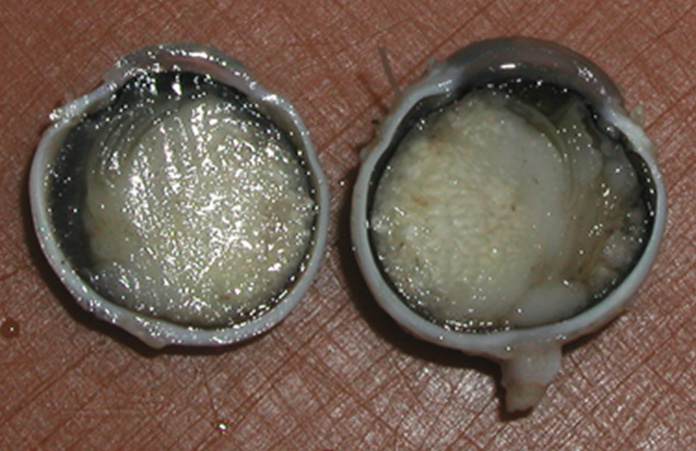 Figure 8: Enucleation due to advanced intraocular retinoblastoma (KDO FN Brno) |
 Figure 9: Recommended management of retinoblastoma (Nurul I, Health and medicine 2014) |
|||||||