Special section Neuroblastoma
Introduction
The term neuroblastoma is commonly used to refer to a spectrum of neuroblastic tumors including:
- neuroblastoma
- ganglioneuroblastoma
- ganglioneuroma.
Neuroblastoma arises from early neural crest precursors that undergo transformation secondary to genetic or epigenetic events that lead to blocked or aberrant developmental differentiation. The neural crest is a transient multipotent embryologic tissue which migrates out of the neural chord during development. Neuroblasts undergo an epithelial to mesenchymal transition and migrate both ventrally and caudally to form components of many tissues including the branchial arches, cardiac and thoracic vessels, and the sympathetic nervous system, which includes the adrenal glands.
Neuroblastomas are a heterogeneous disease, varying in terms of location, histopathologic appearance, and biologic characteristics. Neuroblastoma is most remarkable for it´s broad spectrum of clinical behavior, which can range from spontaneous regression, to maturation to a benign ganglioneuroma, or aggressive disease with metastatic dissemination leading to death.
Epidemiology
It is the third most common childhood cancer (after leukemia and brain tumors) and is the most common solid extracranial tumor in children.
Neuroblastomas are the most common extracranial solid malignant tumor diagnosed during the first two years of life, and the most common cancer among infants younger than 12 months, in whom the incidence rate is almost twice that of leukemia (58 versus 37 per one million infants).
Familial neuroblastoma - although the majority of neuroblastomas are sporadic, in 1 to 2 percent of cases, there is a family history of neuroblastoma. These cases appear to be inherited in an autosomal dominant pattern with incomplete penetrance and a broad spectrum of clinical behavior. Inherited cases usually present at an earlier age than sporadic cases (mean age 9 versus 17 months), and a large proportion have bilateral adrenal or multifocal disease.
Clinical presentation
Neuroblastomas can arise anywhere throughout the sympathetic nervous system:
- the adrenal gland is the most common primary site( 40 percent)
- followed by abdominal (25 percent)
- thoracic (15 percent)
- cervical (5 percent)
- pelvic sympathetic ganglia (5 percent)
- in approximately 1 percent of cases, a primary tumor cannot be identified.
Neuroblastoma metastasizes to
- lymph nodes
- bone marrow
- cortical bone
- dura
- orbits
- liver
- skin
- pulmonary metastases are uncommon
Symptoms of disease
The presenting symptoms reflect the location of the primary tumor and the extent of metastatic disease, if present. Patients with localized disease can be asymptomatic, whereas children with advanced disease appear ill at presentation, usually with systemic symptoms.
Signs and symptoms of neuroblastoma may include:
- Abdominal mass (retroperitoneal or hepatic)
- Abdominal pain or constipation
- Proptosis
- Periorbital ecchymoses ("raccoon eyes", from periorbital ecchymosis caused by orbital metastases)
- Horner syndrome (miosis, ptosis, anhidrosis)
- Localized back pain, weakness (from spinal cord compression)
- Scoliosis, bladder dysfunction
- Palpable nontender subcutaneous nodules
- Opsoclonus myoclonus ataxia syndrome
- Otherwise unexplained secretory diarrhea (from paraneoplastic production of vasoactive intestinal polypeptide [VIP])
- Systemic symptoms (fever, weight loss)
- Bone pain
- Anemia
- Heterochromia iridis (different colors of the iris or portions of the iris)
- Hypertension
Abdominal tumors
Approximately two-thirds of primary neuroblastomas arise in the abdomen; majority in the adrenal gland. Abdominal tumors can present with abdominal pain or fullness, abdominal mass, or rarely intestinal obstruction.
An abdominal mass may be detected in an asymptomatic child by the primary care physician during a routine examination. The mass is typically, but not always, nontender, fixed, and firm.
Thoracic tumors
Primary thoracic tumors may be detected incidentally on radiographs obtained in the evaluation of other complaints (eg, respiratory symptoms, trauma). Mediastinal tumors can cause tracheal deviation or narrowing with resultant stridor, although high thoracic and cervical masses may be associated with a Horner syndrome (ptosis, miosis, and anhidrosis).
Paravertebral tumors
Because neuroblastomas may arise from paravertebral sympathetic ganglia (paraganglia), primary tumors (usually in the retroperitoneum or mediastinum) are able to invade the spinal canal through the neural foramina, creating a so-called dumbbell tumor. The subsequent epidural spinal cord compression, an oncologic emergency, can cause pain, motor or sensory deficits, or loss of bowel and/or bladder control.
Involvement of the cervical paravertebral sympathetic chain and inferior cervical (stellate) ganglion can result in the Horner syndrome (ipsilateral ptosis, miosis, and anhidrosis).
Paraneoplastic syndromes
Several unique paraneoplastic syndromes can be associated with both localized and disseminated neuroblastomas.
- Hypertension – as neuroblastoma is derived from neural crest cells that are destined to form components of the peripheral sympathetic nervous system, that frequently produce catecholamines. Degradation of norepinephrine, epinephrine, and dopamine lead to the final end products VMA (vanillylmandelic acid) and HVA (homovanillic acid). Elevated levels of VMA and HVA can be detected in the serum and urine of approximately 70 to 90 percent of patients with neuroblastoma.
- Opsoclonus-myoclonus-ataxia (OMA) is a paraneoplastic syndrome that occurs in 1 to 3 percent of children with neuroblastoma. Almost 50 percent of children with OMA have an underlying neuroblastoma; neurologic symptoms precede tumor diagnosis in about half of these. The disorder is believed to have an autoimmune pathogenesis. The characteristic symptoms of OMA are rapid, dancing eye movements, rhythmic jerking (myoclonus) involving limbs or trunk, and/or ataxia.
- Secretion of VIP — Autonomous tumor secretion of vasoactive intestinal peptide (VIP) is another paraneoplastic syndrome that is rarely associated with neuroblastoma. VIP secretion can cause abdominal distension and intractable secretory diarrhea with associated hypokalemia.
Metastatic disease
Neuroblastoma metastasizes by both lymphatic and hematogenous routes.
- Regional lymph node involvement is found in 35 percent of children who have apparently localized disease. Involvement of lymph nodes outside the cavity or region of origin (ie, abdomen, thorax, pelvis) is considered to represent disseminated disease.
- Metastatic involvement of the liver is common in infants, and can cause respiratory compromise.
- Metastatic spread to the bones and bone marrow can cause pain (especially with ambulation), blood count abnormalities, and fever. In young children, who cannot complain of pain, bone pain may manifest as a limp or unexplained irritability. Tumor infiltration of the periorbital bones, typically unilateral, can cause the characteristic periorbital ecchymosis ("raccoon eyes") (Figure 1).
- Metastatic spread to the skin manifests as papules or subcutaneous nodules that can be distributed over the entire body. These lesions are present in approximately one-third of children with congenital neuroblastoma, and are typically described as firm, bluish-red, and nontender. The lesions have a distinctive response to rubbing, characterized by central blanching with a surrounding halo of erythema that persists for 30 to 60 minutes.
Differential diagnosis
When the tumor arises in a suprarenal location, shall be considered:
- Wilms' tumor
- hepatoblastoma
In thoracic and retroperitoneal locations, shall be considered:
- lymphoma
- germ cell tumors
- infection
Metastatic involvement of the bone marrow must be distinguished from:
- lymphoma
- small cell osteosarcoma
- mesenchymal chondrosarcoma
- the Ewing sarcoma family of tumors
- undifferentiated soft-tissue sarcomas such as rhabdomyosarcoma
- leukemia
Diagnosis and staging evaluation
All patients with suspected neuroblastoma should undergo:
- complete history and physical examination
- laboratory evaluations including blood counts serum chemistries, and tests of liver and kidney function
- evaluation of urine or serum catecholamine metabolite levels, vanillylmandelic acid (VMA) and homovanillic acid (HVA), should be obtained to assist in diagnosis and monitoring of disease response, since levels are elevated in more than 90 percent of patients with neuroblastoma
- NSE (neuron specific enolase), ferritin and lactate dehydrogenase (LDH) concentrations may be helpful; they may be elevated initially and can be expected to return to normal during adequate treatment.
Staging workup
The minimum requirements for staging include:
- Bilateral iliac crest bone marrow aspirate and biopsy.
- Bone radiographs and either technetium radionuclide scan or I123-MIBG scan (Figure 2). Among patients with known neuroblastoma, I123 MIBG has a sensitivity of approximately 90 percent.
- Abdominal imaging by CT or MRI scan with three-dimensional tumor measurements (Figure 3).
- Chest radiograph (anteroposterior [AP] and lateral); chest CT or MRI are necessary only if the chest radiograph is positive or if abdominal mass or lymph node disease extend into the chest.
- Head CT or MRI should be considered for patients presenting with proptosis, periorbital ecchymoses or as clinically indicated by symptoms.
Radiologic evaluation
- Ultrasonography (US) is often the initial radiologic study in the evaluation of a child with an abdominal mass, the most common presentation of neuroblastoma. US can usually be obtained quickly and may confirm the presence and location of the mass. However, it should be followed by either CT or MRI.
- CT or MRI of the primary tumor may reveal a heterogeneous mass, possibly containing calcifications. When the mass is adjacent to the spine, an MRI is particularly helpful for evaluation of spinal canal invasion.
- A radionuclide bone scan is a sensitive tool for evaluating metastatic spread to cortical bone.
- MIBG is a chemical analog of norepinephrine that is selectively concentrated in sympathetic nervous tissues such as neuroblastoma. It can be labeled with radioactive iodine and imaged by scintigraphy. The MIBG scan is both sensitive and specific for neuroblastoma, and is recommended at diagnosis and repeat evaluations of the tumor.
Pathology
Neuroblastoma is a highly heterogeneous disease, and the pathology varies according to the degree of neural crest differentiation (Figure 4).
The International Neuroblastoma Pathology Classification classifies tumors of neuroblastic origin according to the balance between neural-type cells (primitive neuroblasts, maturing neuroblasts, and ganglion cells) and Schwann-type cells (Schwannian-blasts and mature Schwann cells) into one of three types:
- neuroblastoma
- ganglioneuroblastoma
- or ganglioneuroma.
The degree of differentiation and stromal component of neuroblastoma tumors can be predictive of outcome and is used in the determination of risk category for treatment. According to this system,
Favorable tumors include those that are:
- Poorly differentiated or differentiating neuroblastoma, with low or intermediate mitosis-karyorrhexis index (MKI), patient age ≤ 1.5 years
- Differentiating neuroblastoma and low MKI tumors in patients 1.5 to 5.0 years of age
- Ganglioneuroblastoma, intermixed, regardless of age
- Ganglioneuroma, regardless of age
Unfavorable tumors include those that are:
- Undifferentiated or high MKI tumors in patients of any age
- Poorly differentiated and/or intermediate MKI tumors in patients 1.5 to 5.0 years of age
- Any grade of differentiation and any MKI class in patients ≥5 years of age
- Nodular ganglioneuroblastoma, regardless of age
Prognostic markers
Clinical diversity correlates closely with numerous clinical and biological factors including:
- patient age (the younger the age at diagnosis, the better the survival rate)
- tumor stage
- histology (favourable x unfavourable)
- genetic and chromosomal abnormalities (NMYC, del 1p, 11q LOH; 17q gain,ploidy…)
Most important genetic prognostic marker is the presence of NMYC amplification (Figure 5). High levels of the MYCN protein, a DNA binding transcription factor known to cause malignant transformation in both in vitro and in vivo tumor models. A 50- to 400-fold amplification of MYCN is found in approximately 25 percent of neuroblastomas and is an indicator of poor prognosis.
In additional to structural chromosomal changes, alterations in total DNA content, which presumably result from mitotic dysfunction, are an important indicator of both outcome and response to therapy. Neuroblastomas with a higher DNA content (hyperdiploid, with a DNA index [DI] >1) are associated with lower tumor stage, better response to initial therapy, and an overall better prognosis than diploid tumors (ie, DI = 1), particularly if they lack MYCN amplification.
The molecular and cytogenetic characterization of neuroblastomas is a routine part of the clinical evaluation because of the influence of these findings on clinical outcome. The selection of treatment is mostly based upon presence or absence of molecular and genetic factors.
Treatment and prognosis of neuroblastoma
The modern treatment of neuroblastoma is determined based on risk categories. Patients are classified into:
- low
- Intermediate
- and high risk categories.
based on the following characteristics at the time of diagnosis:
- Stage of the disease
- Patient age
- Histologic appearance of the tumor
- Presence or absence of amplification of the MYCN oncogene
- Quantitative DNA content of the tumor (DNA index or ploidy).
Low-risk disease
- In general, tumor outcomes for children with low-risk neuroblastoma are excellent.
- Surgery is the mainstay of treatment for low risk tumors although some infants and children need additional chemotherapy and others can be observed without surgery.
- Observation — Expectant observation is a reasonable alternative in some newborns with small adrenal masses.
Intermediate-risk
The standard approach for these patients includes
- Moderately intensive multiagent chemotherapy with resection when possible.
- Radiation therapy is rarely indicated but should be considered for those with tumor progression or tumor-related life threatening or organ-threatening complications unresponsive to chemotherapy.
- The four-year EFS and overall survival rates for patients with normal MYCN and favorable histology was 100 percent each, while for infants with at least one unfavorable biologic feature, they were 90 and 93 percent, respectively.
High-risk disease
- Patients at the highest risk for disease progression and mortality are those who are older than 18 months year of age and have disseminated disease, or localized disease with unfavorable markers such as MYCN amplification.
- Despite aggressive multimodality therapy, current survival rates remain unacceptably low (approximately 40 percent), and the improved outcomes have come at a cost of significant early and late toxicity.
Author: Pavel Mazánek, MD
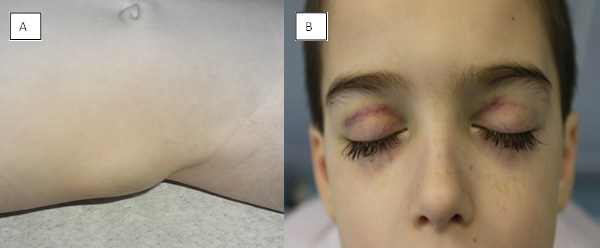 Figure 1: Metastasis of neuroblastoma: (A) skin, (B) orbits – „racoon eyes“ |
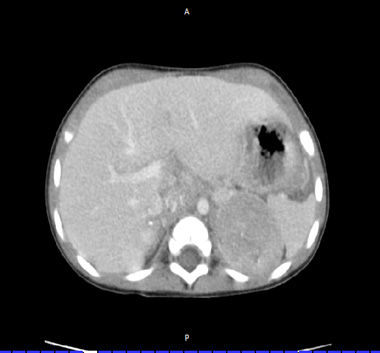 Figure 2: Abdominal CT scan, neuroblastic tumor of the left adrenal gland |
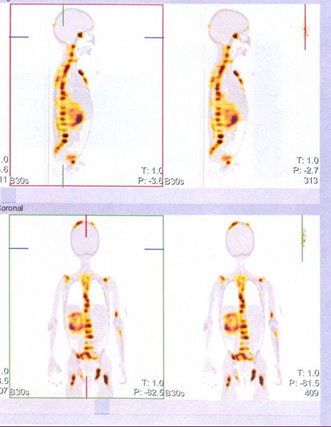 Figure 3: I-123 MIBG scans of a patient with multiple bone metastases of the spine, sternum, skull, long bones of the upper and lower extremities. The primary tumor is in the right adrenal gland. |
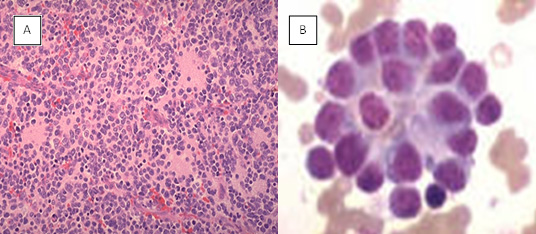 Figure 4: Neuroblastoma. (A) typical “small blue round cell tumor” of a childhood, (B) rossetes of neuroblastic cells |
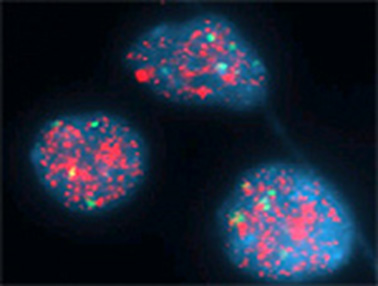 Figure 5: NMYC amplification in neuroblastoma; laboratory method – FISH (fluorescent in situ hybridisation). Normally we should see 2 green control signals (centrosomic probe on chromosome 2) and 2 pink signals (NMYC). |
|||