Special section Renal tumors – Wilms tumor (nephroblastoma)
Definition
Tumors of the kidneys represent about 4–6 % of all malignant diseases in children up to 15 years of the age.
Tumors origin in renal parenchyma:
- nephroblastoma (Wilms tumor)
- clear cell sarcoma (CCSK)
- rhabdoid tumor of the kidney (RTK)
- congenital mesoblastic nephroma (CMN) (Figure 1)
Kidney can be affected (or infiltrated) also by metastatic spread of other malignant processes (leukemia, lymphoma, sarcoma).
The most common form of primary pediatric renal cancer is nephroblastoma (Wilms tumor), which represent about 87 % of all renal cancer in children. It is a typical embryonal tumor origin from primitive metanefrogenic blastoma and it is the 5th most common childhood cancer.
Other renal tumors in childhood are quite rare and only 4% of all kidney tumors are benign. Incidence of different types of renal tumors is age specific (Figure 2).
Nephroblastoma (Wilms tumor) was first described in 1899 by surgeon Max Wilms.
Epidemiology
- Annual incidence is 1 : 10 000 in children younger than 15 years. A lower incidence has been noted in Asian countries (Japan, India, Singapore), a higher incidence is found in Scandinavia, Brazil and black children (Africa)
- represents 87 % of pediatric renal tumors
- age peak is up to 5 years of the age (average age at diagnosis is 3.5 year), it becomes less common in older children after the age of 7 – 10 years
- slight female predominance ( M : F ratio is 0.92 . 1)
- usually is unilateral, only 5 – 10 % of Wilms tumors are bilateral with both kidneys involvement
- extremely rare is Wilms tumor in adult population (less than 3% of all cases of Wilms tumor)
Etiology and etiopathogenesis
Causes of nephroblastoma are mostly unknown with sporadic occurrence.
There is no association with environmental factors, either during the mothers pregnancy or after delivery.
1–2% of Wilms tumor are familial, with at least one relative also diagnosed with Wilms tumor (siblings, uncle, cousin …). Analyses of affected families did not detect any specific genetic mutation, therefore predisposition due to an autosomal dominant mutation with incomplete penetrance or multigene model is supposed to cause familial Wilms tumor occurrence.
12–15% of Wilms tumor is associated with congenital birth defects and anomalies (anomalies of genitourinary tract – hypospadia, cryptorchidism, horseshoe kidney), aniridia or hemihypertrophy (Figure 3).
Wilms tumor is strongly associated with several genetic cancer predisposition syndromes in 10–15 % of cases. Children with inherited germ line mutations and syndromes develop tumors at an earlier age and bilateral cases of Wilms tumor are more frequent (Figure 4).
Three candidate gene loci are identified in genetics of Wilms tumor:
-
WT1 gene (chromosome 11p13):
- WAGR syndrome (gene deletion) (Figure 5)
- Denys Drash syndrome (point mutation)
- Frasier syndrome (point mutation)
- paternal gene is “silencing”
- WT2 gene (chromosome 11q15):
- Beckwith-Wiedemann syndrome (deletion)
- paternal gene is not “silencing”
Other syndromes associated with Wilms tumor:
- Bloom syndrome (Figure 6)
- Perlman syndrome
- Li Fraumeni syndrome
Some somatic mutations have been detected in Wilms tumor samples presumably contribute to the pathogenesis of the tumor:
- WTX mutation in 20–30% tumors
- CTNNB1 (β catenin) in 15% tumors
- TP53 in 5% tumors
Clinical presentation (symptoms)
The most common initial presentation of Wilms tumor is an asymptomatic palpable (or visible) abdominal usually painless tumor mass, hard, fixed (Figure 7).
Incidental finding is quite common (during the bathing or dressing the child).
General condition of the child is not altered.
Non-specific symptoms are present in less than 1/3 children:
- subfebrility, weakness, pallor,
- loss of appetite
- constipation, abdominal pain
10–30 % patients develop micro or macroscopic hematuria (sometimes only transient) due to tumor penetration and involvement of renal pelvis.
Renovascular hypertension (high blood pressure) is variable, has been reported in 25 % of children, caused by paraneoplastic production of renin by tumor cells.
Rarely Wilms tumor may arise outside the kidney (retroperitoneum).
The most common sites of metastases are the lungs and liver, non hematogenous extension includes lymph nodes involvement and extension into the renal vein and inferior vena cava.
Diagnostic procedures
Diagnostic work-up begins with family and personal history (family history of cancer, abortions, malformations or congenital anomalies)
Physical examination (very gentle palpation of the abdomen – risk of tumor rupture !), movement of the mass during respiration, looking for signs of overgrowth, peripheral lymph nodes, edemas, hypertension, passing stool/urine, macroscopic hematuria.
Imaging studies (Figure 8 and 9):
- abdominal ultrasound (+ doppler to exclude tumor thrombus in inferior vena cava) (Figure 10)
- abdominal CT scan (Figure 11, 12 and 13)
- MRI of the abdomen provide more detailed images of the kidney than CT, don´t expose child to radiation, but unfortunately it takes longer than CT and examination should be done under general anesthesia (GA) in younger children (Figure 14)
- chest XC-ray + CT scan of the lung (Figure 15)
- CT scan of the brain is indicated only in rhabdoid tumor of the kidney bone scan (scintigraphy) is reserved for patients with CCSK 18 FDG PET scan is not a part of routine diagnostic work up
Scintigraphy of the kidneys (DTPA) is important before surgery to see the function of the both kidneys (involved by tumor as well as contralateral kidney)
Lab tests:
- hematology ( CBC, coagulation profile)
- biochemistry (ionogram, urea, creatinin, uric acid, cystatin C, LDH)
- urine analysis (to exclude micro/macroscopic hematuria)
- there is no specific tumor marker available for Wilms tumor
Histolopathology (including immunohistochemistry): nephroblastoma covered a large spectrum of special variants that digger in morphological features, natural history and prognosis (Figure 16):
- classic triphasic embryonal tumor recapitulates the normal development of the kidney with components of blastema, stroma and epithelial tubules and glomeruli, but one element may predominate and may have prognostic relevance
- anaplastic (diffuse or focal anaplasia)
- blastemal
- stromal
Cytogenetic and molecular genetic studies are currently essential for better characterization of clinical behavior of the tumor, for risk stratification and appropriate treatment. There are using as predictive and prognostic biomarkers. The role of biomarkers:
- to predict response to chemotherapy
- direct impact to prognosis
- key to understand tumorigenesis
Primary molecular genetic changes with predisposition to develop Wilms tumor:
- WT1 gene - t(11;13)
- WT2 gene - t( 11;15)
Secondary (epigenetic) changes associated with malignant progression (Figure 17)
- somatic mutation TP53 gene
- somatic mutation CTNNB1
- somatic mutation WTX
- 16qLOH, 1pLOH, gain 1q, 6, 8, 12
- overexpression CD44
- LOH IGF2 (potent growth factor)
Differential diagnosis
For differential diagnosis of renal masses in childhood is essential to exclude (Figure 18):
- Congenital anomalies mimicking tumor (renal cysts, horseshoe kidney ……)
- inflammatory lesions ( abscess, mycotic lesions, inflammatory pseudotumor)
- benign tumors (mesoblastic nephroma, fibroma, angiomyolipoma, adenoma, teratoma)
- other malignant tumors (renal cell carcinoma, PNET, fibrosarcoma, clear cell sarcoma, rhabdoid tumor of the kidney)
- metastases to the kidney ( specially hematology malignancies –leukemia, lymphoma)
- nephroblastomatosis (Figure 19 and 20)
Also is crucial to exclude other pediatric embryonal abdominal tumors (neuroblastoma, hepatoblastoma, rhabdomyosarcoma, germ cell tumor) and in older children B-cell non Hodgkins lymphoma.
Therapy
Treatment of Wilms tumor is multimodal with interdisciplinary cooperation. Stage and risk adapted approach is the standard of care.
Classification and risk groups are based on individual risk assessment
- histopathology and tumor type (Figure 21)
- age of the patient ( < 2 years >)
- size/volume of the tumor (550gr or 500ml)
- stage of disease (Figure 22)
- biology characteristics of the tumor (cytogenetics)
Treatment modalities
Chemotherapy:
-
SIOP (Europe)
- 4 weeks neoadjuvant chemotherapy (vincristin, actinomycin D = AV)
- delayed surgery
- adjuvant chemotherapy according to risk group
- COG (USA) primary surgery followed by adjuvant chemotherapy according to risk group
Surgery:
- transperitoneal nephrectomy through transverse abdominal incision with peroperative inspection of contralateral kidney and regional lymph node sampling (Figure 23)
-
heminephrectomy (nephron sparing surgery) - very precise indications:
- solitary kidney
- horseshue kidney
- bilateral Wilms tumor
- Wilms tumor associated with genetic syndrome
- small tumor (<2cm) localized at the pole of the kidney
Radiotherapy:
Due to risk of the late consequences indications for radiotherapy have been reduced. Radiotherapy is indicated in case of:
- non radical surgery, ruptura of the tumor (spontaneous or peroperative) and intraabdominal spillage of the tumor
- lymph node involvement, persistent tumor trombus = stage III
- residual lung metastases after neoadjuvant chemotherapy
Bilateral Wilms tumor:
- patients with synchronous bilateral Wilms tumor should not undergo initial radical nephrectomy
- after neoadjuvant chemotherapy (at least 6 weeks) nephron sparing surgery should be performed, the kidney with lower burden is addressed first
- radiotherapy is contraindicated due to high risk of renal failure
Prognosis and outcome
Wilms tumor is a curable type of childhood cancer
Factors influencing prognosis (Figure 24):
-
histology of tumor:
- favorable
- unfavorable histology: diffuse anaplasia, blastema
- response to neoadjuvant chemotherapy (% of necrosis, regressive changes)
- clinical stage (localized or metastatic disease)
- age of the child at the time of diagnosis
- weight and volume of the tumor
- cytogenetic features: unfavorable is translocation 1pLOH,16qLOH
Dramatic improvement of survival has been achieved within the last 20 years in children with Wilms tumor (Figure 25).
4-year overall survival is more than 90 % in localized disease and over 85% in advanced disease for favorable histology and about 83% in localized unfavorable histology. Treatment results of advanced unfavorable histology remains unsatisfied (Figure 26).
Author: Viera Bajčiová, MD, PhD
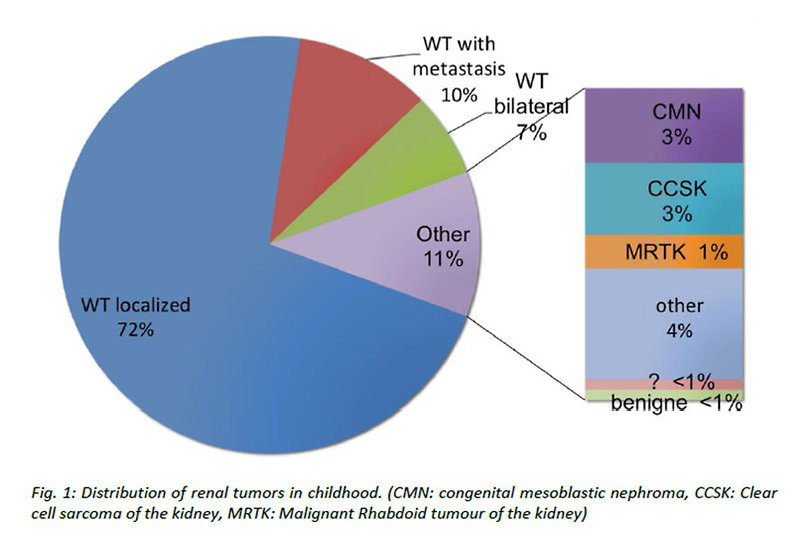 Figure 1: Distribution of renal tumors in childhood |
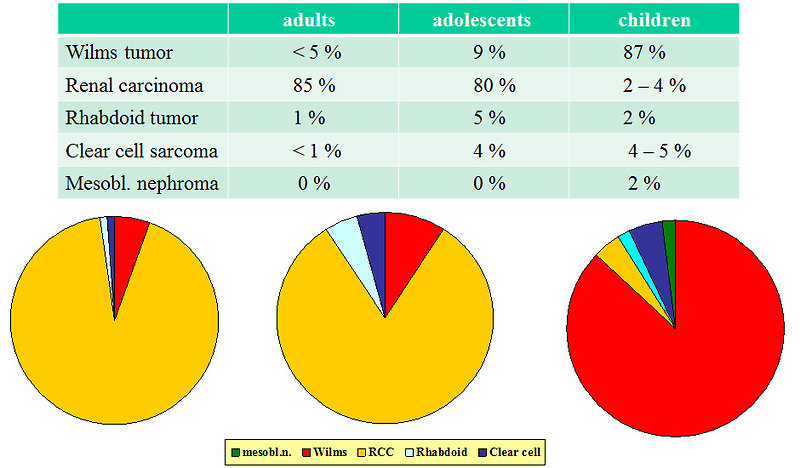 Figure 2: Age specific distribution of renal tumors |
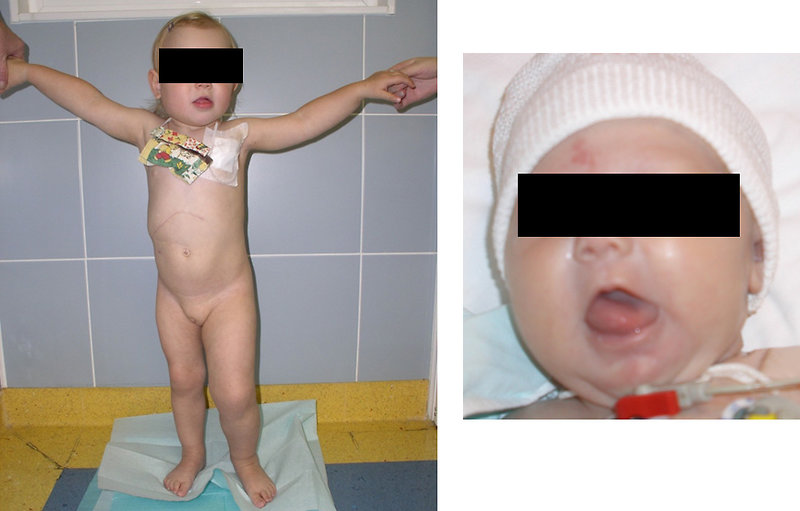 Figure 3: Hemihypertrophy |
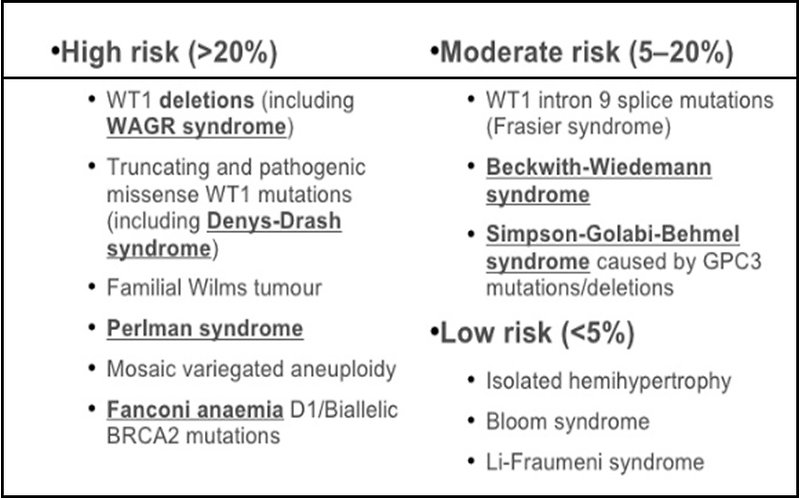 Figure 4: Association of Wilms tumor with genetic syndromes |
 Figure 5: WAGR syndrome |
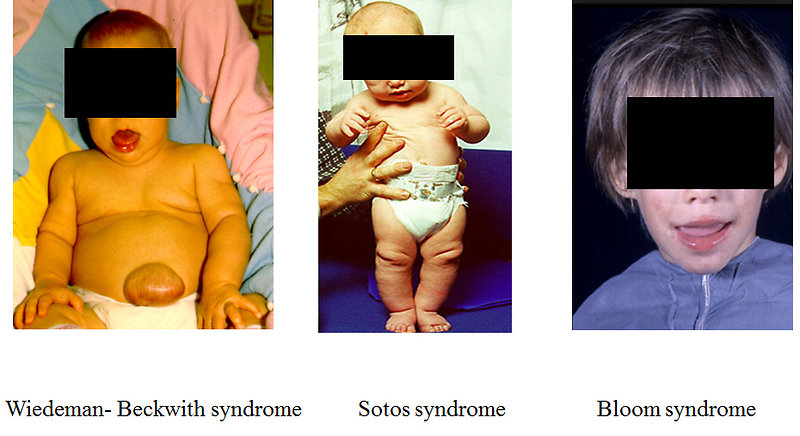 Figure 6: Genetic predisposition syndromes associated with Wilms tumor |
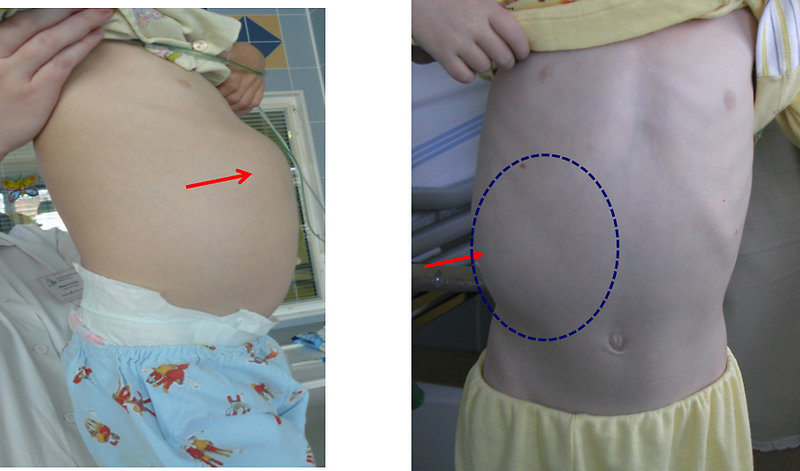 Figure 7: Clinical presentation of Wilms tumor of the right kidney |
 Figure 8: Flow diagram for initial diagnostic work up |
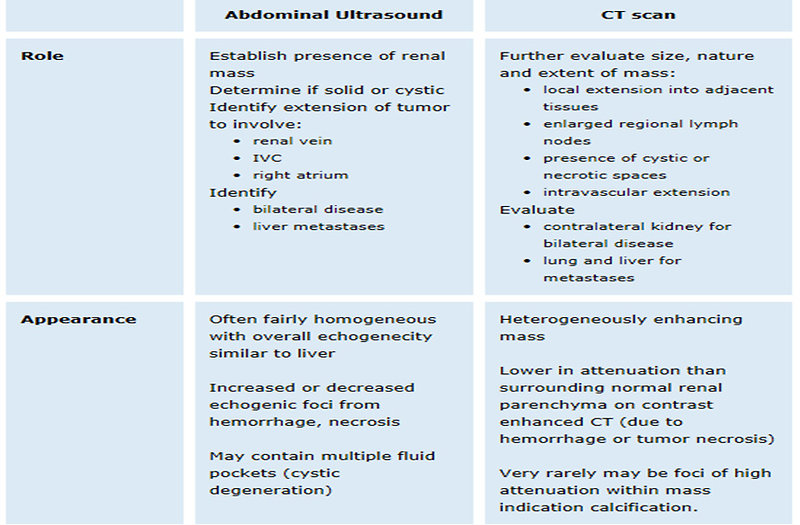 Figure 9: Radiology findings of renal tumors |
 Figure 10: Radiology - UZ and CT |
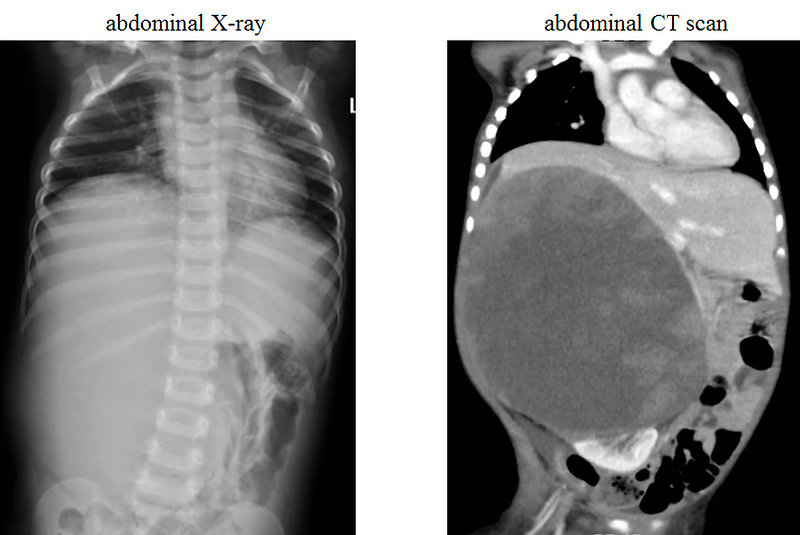 Figure 11: X-ray and CT of Wilms tumor |
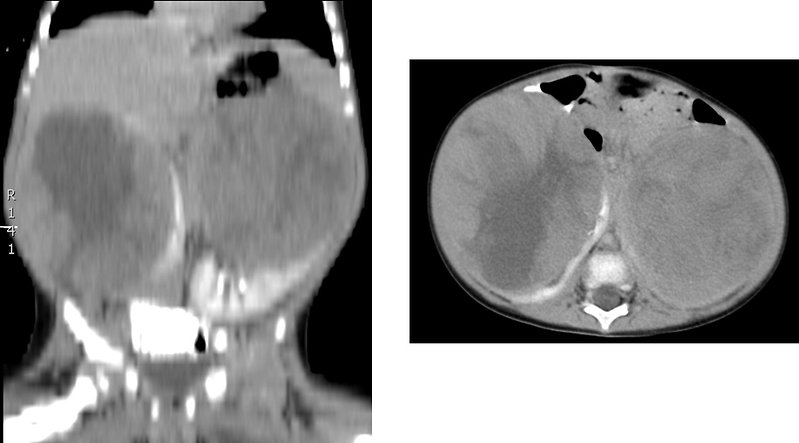 Figure 12: Bilateral Wilms tumor |
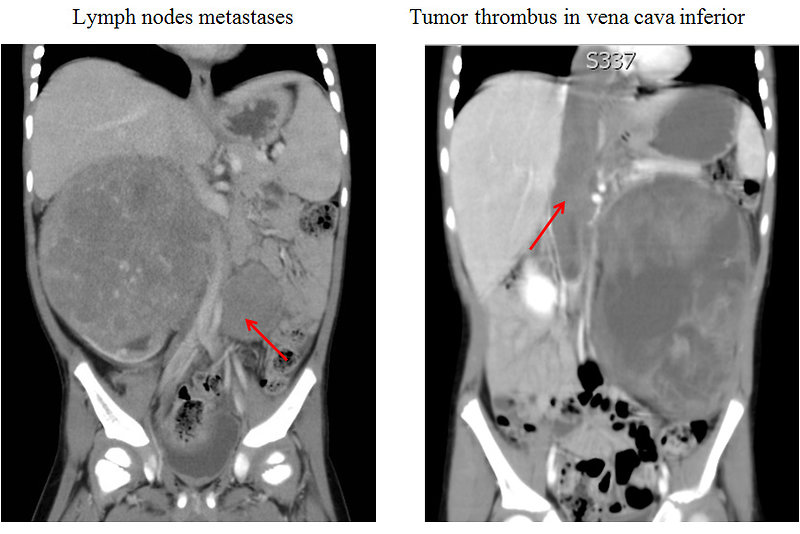 Figure 13: CT scan if metastatic Wilms tumor |
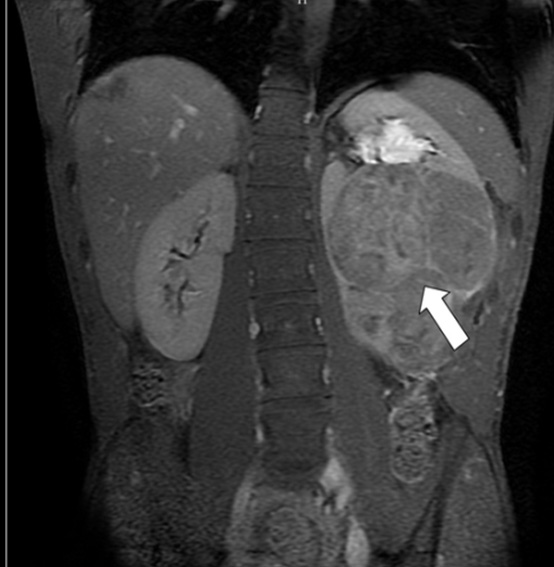 Figure 14: MRI of Wilms tumor |
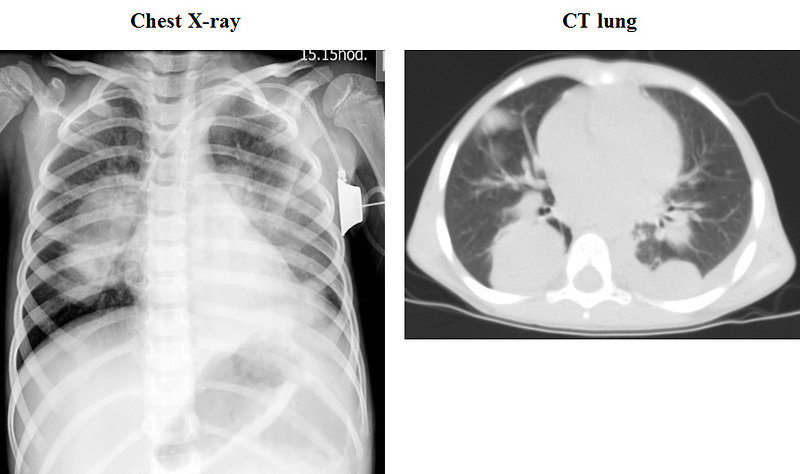 Figure 15: Lung metastases of Wilms tumor |
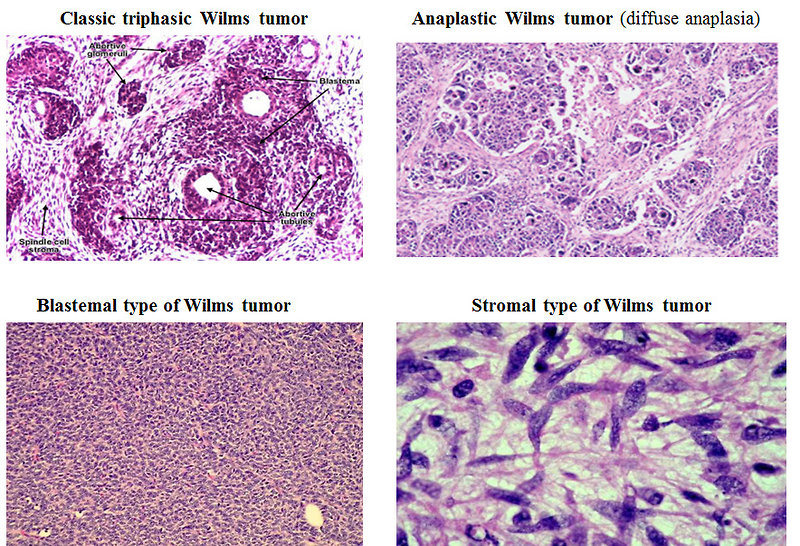 Figure 16: Pathology – types of WT |
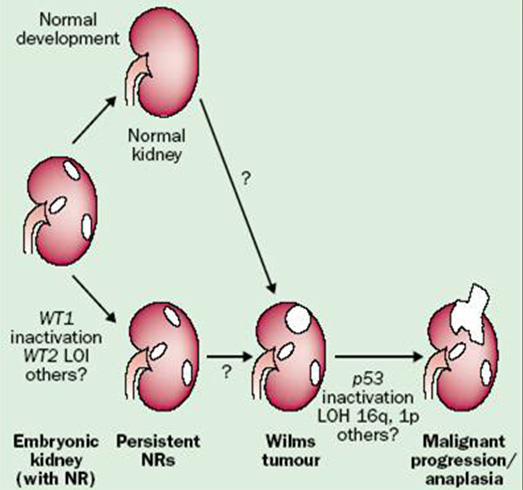 Figure 17: The role of genetic and epigenetic changes in tumorigenesis of Wilms tumor |
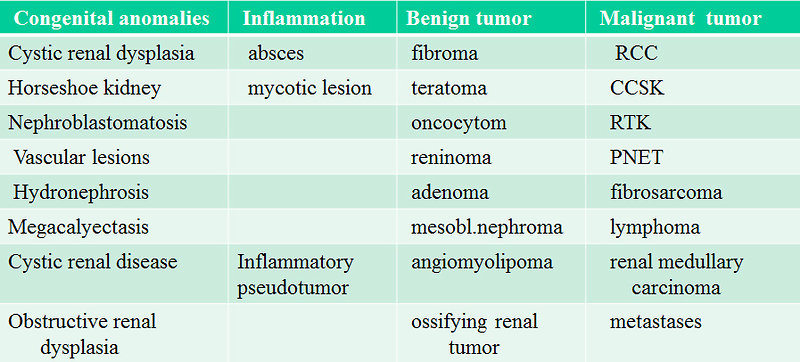 Figure 18: Differential diagnosis of pediatric renal masses |
 Figure 19: Nephroblastomatosis (nephrogenic rests) |
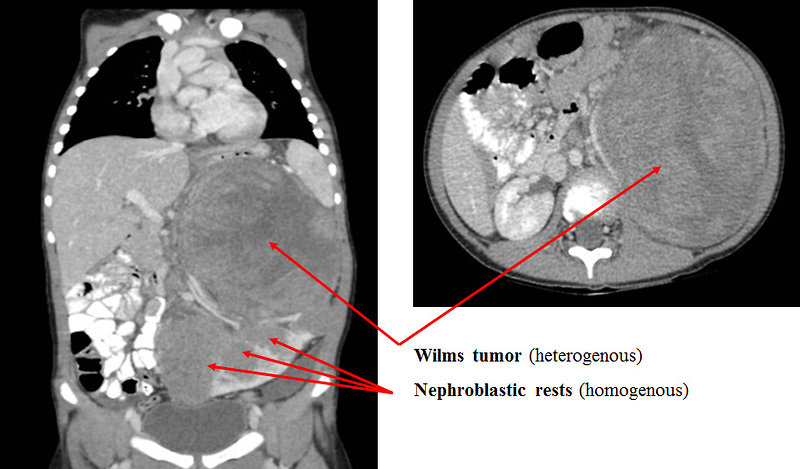 Figure 20: CT scan – Wilms tumor arising from perilobar nephroblastomatosis |
 Figure 21: Pathology working classification of childhood renal tumors |
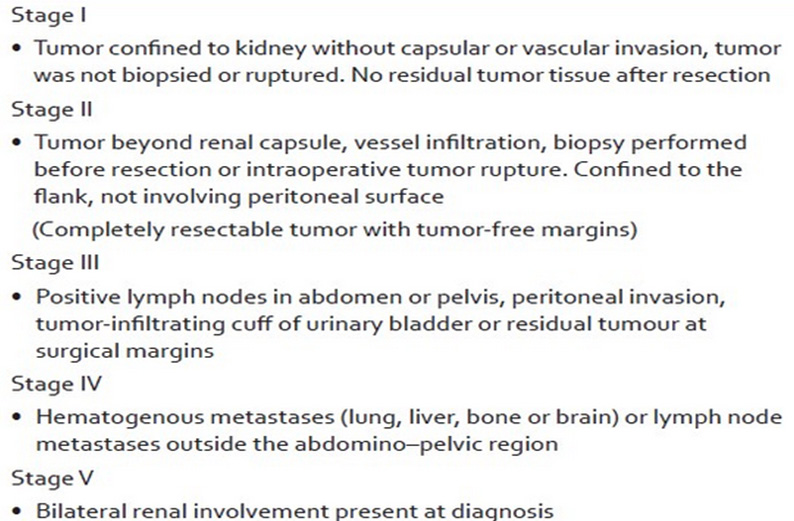 Figure 22: Clinical staging of Wilms tumor |
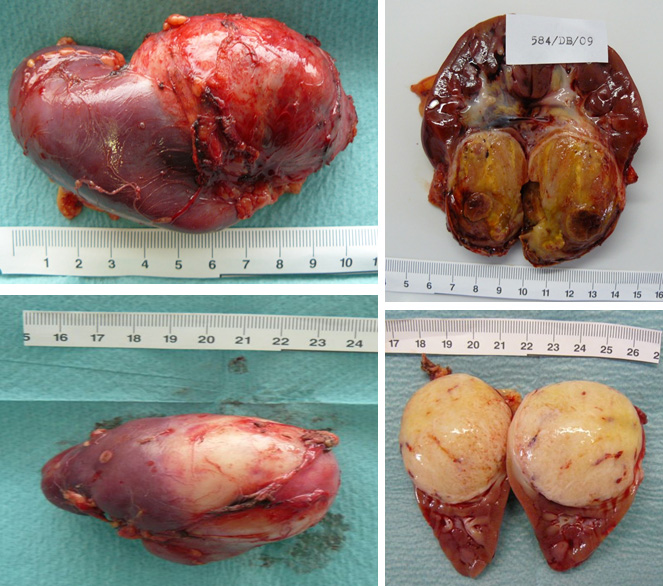 Figure 23: Wilms tumor after nephrectomy |
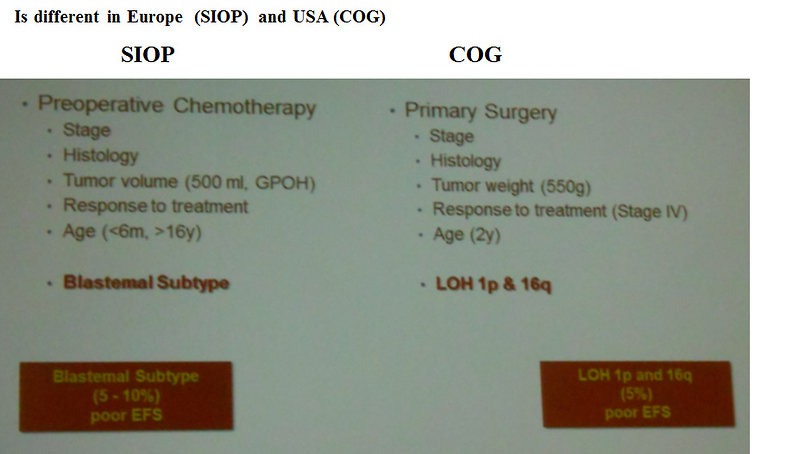 Figure 24: Risk group stratification of Wilms tumor |
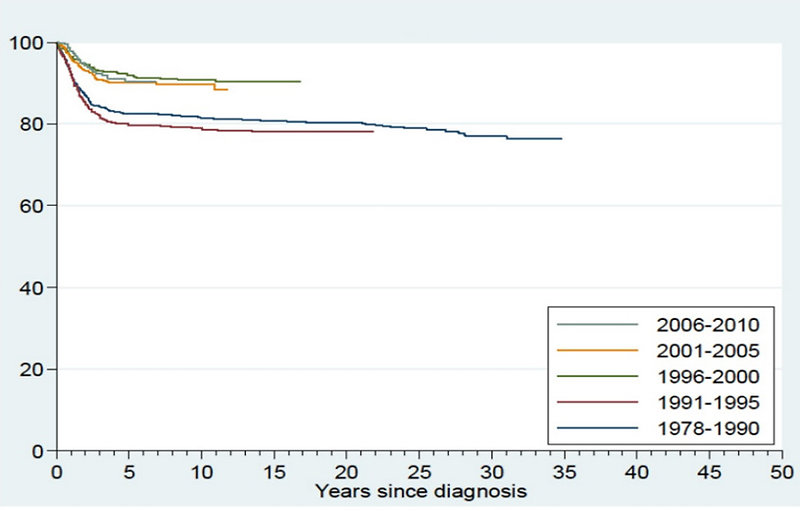 Figure 25: Improvement in survival of Wilms tumor |
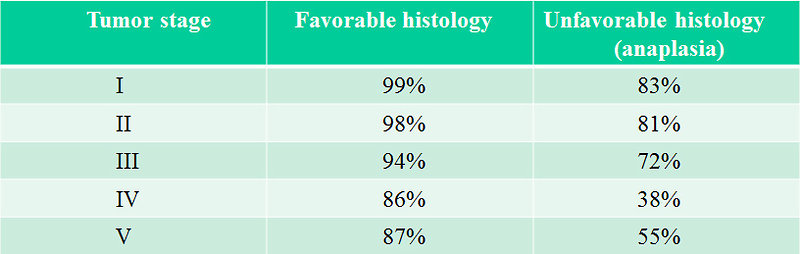 Figure 26: 4-year survival rate of Wilms tumor |
||||||