Special section Acute lymphoblastic leukemia
Definition
Acute lymphoblastic leukemia (ALL) is a malignant (clonal) disease of the bone marrow in which early lymphoid precursors proliferate and replace the normal hematopoietic cells of the marrow.
Epidemiology
- Acute lymphoblastic leukemia (ALL) is the most common type of cancer and leukemia in children.
- ALL accounts for 26% of all cancers in children up to 14 years of age, and for 75% of pediatric leukemia cases.
- ALL is most common in childhood, with a peak incidence at 2–6 years of age.
- In adults, ALL is less common than acute myelogenous leukemia (AML).
- ALL is slightly more common in males than in females.
- The survival rate of childhood ALL is approaching 90%, although only 20-40% of adults are cured.
Classification
Prior to 2008, subtyping of all acute leukemias (including acute myelogenous leukemia, AML) used the French-American-British (FAB) classification, in which ALL was classified as (Figure1):
- ALL-L1: small uniform cells
- ALL-L2: large varied cells
- ALL-L3: large varied cells with vacuoles (bubble-like features)
In 2008, the World Health Organization classification identified three therapeutically distinct categories. These are identified by immunophenotyping of surface markers of the abnormal lymphocytes. This subtyping helps determine the prognosis and the most appropriate treatment in treating ALL. It is substantially amplified by cytogenetics and molecular diagnostics tests:
-
Acute lymphoblastic leukemia/lymphoma (Synonyms: Former FAB L1/L2)
-
Precursor B acute lymphoblastic leukemia, cytogenetic subtypes:
- t(12;21)(p12,q22) TEL/AML-1
- t(1;19)(q23;p13) PBX/E2A
- t(9;22)(q34;q11) ABL/BCR
- T(V,11)(V;q23) V/MLL
- Precursor T acute lymphoblastic leukemia/lymphoma
-
Precursor B acute lymphoblastic leukemia, cytogenetic subtypes:
- Burkitt's leukemia/lymphoma (Synonyms: Former FAB L3)
- Biphenotypic acute leukemia
Signs and symptoms
Signs and symptoms of ALL are mostly non-specific and include the following:
- Fever
- Decreased neutrophil count
- Signs and symptoms of anemia, such as pallor, fatigue, dizziness, palpitations, cardiac flow murmur, and dyspnea with even mild exertion
- Bleeding (eg, from thrombocytopenia due to marrow replacement)
- Disseminated intravascular coagulation (DIC) at diagnosis (about 10% of cases)
- Palpable lymphadenopathy
- Symptoms related to a large mediastinal mass (eg, shortness of breath), particularly with T-cell ALL
- Bone pain (severe and often atypical)
- Left upper quadrant fullness and early satiety due to splenomegaly (about 10-20% of cases)
- Symptoms of leukostasis (eg, respiratory distress, altered mental status)
- Renal failure in patients with a high tumor burden
- Infections, including pneumonia
- Petechiae (particularly on lower extremities) and ecchymoses
- Signs relating to organ infiltration with leukemic cells and lymphadenopathy
- Rashes from skin infiltration with leukemic cells
Diagnosis
Morphological identification of lymphoblasts by microscopy and immunophenotypic determination of lineage commitment and developmental stage by flow cytometry are essential for correct diagnosis of ALL
Other laboratory tests and studies used in the workup for ALL include the following:
- Complete blood count with differential
- Coagulation studies
- Peripheral blood smear
- Chemistry profile, including lactic dehydrogenase, uric acid, liver function studies, and BUN/creatinine
- Chest x-ray
- Multiple-gated acquisition scanning
- Bone marrow aspiration and biopsy (definitive for confirming leukemia)
- Flow cytometry
- Cytogenetics and molecular genetic analysis
Differential diagnoses
- Acute Myelogenous Leukemia
- Non-Hodgkin Lymphoma
- Disseminated solid tumors (neuroblastoma, rhabdomyosarcoma)
- Infections (leukemoid reaction)
Etiology
It has been known for several decades that the majority of childhood ALL cases harbour gross chromosomal alterations. In ALL, these include:
- high hyperdiploidy with non-random gain of at least five chromosomes (including X, 4, 6, 10, 14, 17, 18, and 21)
- hypodiploidy with fewer than 44 chromosomes
- recurring translocations, including
- t(12;21) encoding TEL-AML1
- t(1;19) encoding E2A-PBX1
- t(9;22) encoding BCR-ABL1
- MLL rearrangement involving 11q23 with a wide range of partner genes.
These alterations are of key importance in both the pathogenesis and clinical management of ALL. Many of these chromosomal rearrangements disrupt genes that regulate normal hematopoiesis and lymphoid development, activate oncogenes or constitutively activate tyrosine kinases (eg, ABL1).
Several of these alterations are significantly associated with outcome, particularly in B-ALL, and are used in risk stratification.
Prognosis
Progress in the treatment of acute lymphoblastic leukemia in children and adolescents has been made in the last 10 to15 years mainly through refinement of risk stratification and adaptation of chemotherapy.
The advanced knowledge about genetics of ALL and molecular regulation of treatment response and resistance represents the basis for the design of contemporary treatment protocols.
Risk factors/risk assignment
Based on the response to therapy and distinct clinical, biological and genetic alterations, patients with ALL are divided into three prognostic groups:
- good (standard) risk
- intermediate risk
- and high risk.
Clinical and biological prognostic factors
- Age (infant or ≥10 years old) presenting leukocyte count (≥50×109/L) race (Hispanic or black), male sex, and T-cell immunophenotype have been considered adverse clinical prognostic factors in children,
- Infants with MLL rearrangement, especially those < 6 months old with a leukocyte count >300×109/L at diagnosis, still have a dismal prognosis.
Prognostic genetic alterations in ALL
Several genetic alterations (see Etiology) are significantly associated with outcome, particularly in B-ALL, and are used in risk stratification.
As cytogenetic alterations alone do not accurately predict the risk of relapse, there is great interest in the relation between novel genetic alterations and ALL outcome. The most consistent association is that of IKZF1 alterations and poor outcome. IKZF1 gene encodes IKAROS transcription factors, and is required for lymphoid lineage development . IKZF1 is altered in 15% of B-ALL cases and is a hallmark of high-risk ALL subtypes.
Response to therapy an ALL prognosis
Early treatment response is predictive of the risk of relapse and is used to assign patients to subsequent risk-adapted therapy. Methods that track residual leukemic cells by flow cytometry (detecting aberrant immunophenotypes) and by PCR amplification (detecting leukemia-specific immunoglobulin and T-cell receptor genes or fusion transcripts) allow the recognition of ALL cells present at levels well below those detectable by microscopic morphologic assessment, ie, minimal residual disease (MRD). MRD is currently the most powerful prognostic indicator in childhood and adult ALL.
Treatment
- The earlier ALL is detected, the more effective the treatment.
- The aim of the therapy is to induce a lasting remission, defined as the absence of detectable cancer cells in the body (usually less than 5% blast cells in the bone marrow).
Treatment of ALL typically spans 2–2.5 years, comprising 3 phases: remission-induction, consolidation and continuation (or maintenance) (Figure 2):
Remission-induction therapy
Four to 6 weeks of remission-induction treatment eradicates the initial leukemic cell burden and restores normal haematopoiesis in 96–99% of children and 78–92% of adults. The chemotherapy agents typically include a glucocorticoid (prednisone or dexamethasone), vincristine, and asparaginase, with or without anthracycline.
Consolidation chemotherapy
Consolidation therapy is administered after remission-induction to eradicate residual leukemic cells.This phase commonly uses high-dose (ie, 1–8g/m2) methotrexate (MTX) with merkaptopurine.
Maintenance chemotherapy
The aim of maintenance therapy is to kill any residual cell that was not killed by remission induction and intensification regimens. Continuation therapy typically lasts up to 2 years from diagnosis or longer and comprises mainly daily mercaptopurine and weekly methotrexate.
Central nervous system (CNS) therapy
Since ALL cells sometimes penetrate the CNS, most protocols include delivery of chemotherapy into the CNS fluid (termed intrathecal chemotherapy). CNS therapy begins early during induction and continue during the consolidation/intensification and maintanance therapy period. The rationale is based on the presence of CNS involvement in 10%-40% at diagnosis.
Bone marrow transplantation
For well-defined high-risk patients and some patients with relapsed disease, allogeneic hematopoetic stem cell transplantation was also shown to be effective.
Radiation therapy
- Radiotherapy is used as the preparations for a bone marrow transplant (total body irradiation).
- Radiation in the form of whole-brain radiation is also used for central nervous system prophylaxis, to prevent recurrence of leukemia in the brain. Whole-brain prophylaxis radiation used to be a common method in treatment of children’s ALL. Recent studies showed that CNS chemotherapy provided results as favorable but with less developmental side-effects
Biological therapy
For some subtypes of relapsed ALL, aiming at biological targets in combination with chemotherapy, has given promising results in clinical trials.
- Ph+ALL is characterised by a reciprocal translocation between chromosomes 9 and 22 (Philadelphia chromosome) that fuses genetic sequences of the bcr gene on chromosome 22 with c-abl sequences translocated from chromosome 9. The BCR-ABL fusion proteins are characterised by a constitutive protein tyrosine kinase (PTK) activity that is absent in the normal ABL protein. This dysregulated PTK activity, which results in changes of multiple signal transduction pathways, is crucial to the transforming activity of the BCR-ABL fusion proteins and their ability to cause leukemias in vivo. Therefore, inhibition of the PTK activity of this oncoprotein is a rational therapeutic approach for BCR-ABL expressing leukemia. The currently used ABL tyrosine kinase inhibitors are IMATINIBand DASATINIB.
- In ongoing clinical trials, a CD19-CD3 bi-specific monoclonal murine antibody, blinatumomab, is showing great promise.
Immunotherapy
Chimeric antigen receptors (CARs) have been developed as a promising therapy for ALL. This technology uses a single chain variable fragment (scFv) designed to recognize the cell surface marker CD19 as a method of treating ALL. CD19 is a molecule found on all B-cells and can be used as a means of distinguishing the potentially malignant B-cell population in the patient.
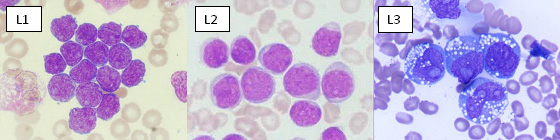 Figure 1: FAB classification of ALL |
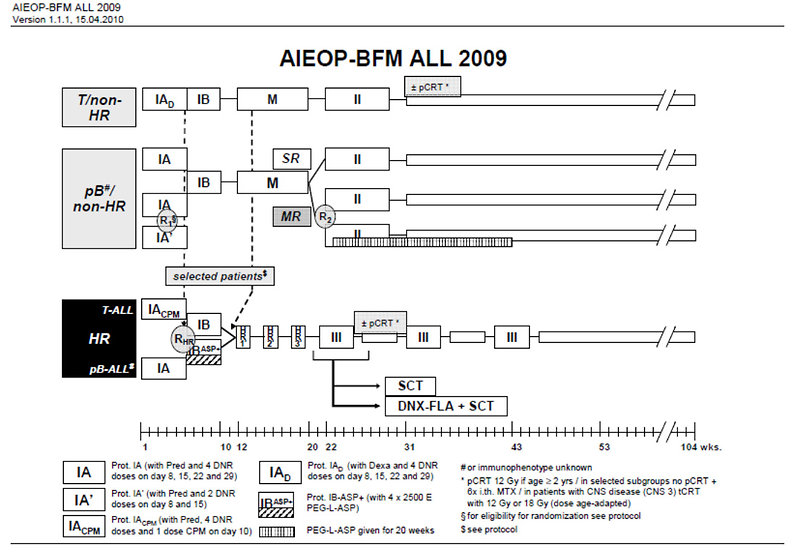 Figure 2: Scheme of international protocol for ALL treatment |
||||||