Special section Rare tumors Adult types of tumors
Introduction
Adult types of tumors are tumors typical for adult and older population and most of them are of epithelial origin (carcinomas). They are very rare in pediatric and adolescent age. Several differencies and similarities between pediatric and adult types of tumors are documented. Close bilateral cooperation between pediatric and adult oncologists is needed for appropriate management of these rare tumors. (Figure 1)
Pediatric tumors vs adult tumors differences:
- Origin of the tumor: most of the adult cancer is of epithelial origin. Pediatric types of tumor are of mesenchymal origin or embryonal tumors arising from undifferentiated tissues.
- Premalignant stage is common in adult cancer, but not for pediatric tumors
- Etiology: environmental factors are fundamental for etiopathogenesis of adult cancer, but have minimal role in etiology of pediatric tumors, etiology of pediatric cancer still remains unknown in most of the cases
- Minimal impact of screening: for pediatric tumors we are not able to define risk groups
- Biology behavior and aggressiveness of pediatric cancer is much higher than adult cancer with rapid growth, early metastatic spread
- Host of the tumor /child differs in many ways from the adult – child is growing and still developing person, but with no comorbidities and excellent regenerative and metabolic processes.
- Therapeutic outcome: is much better for pediatric type of tumors due to their biology behavior. High percentage of cancer cell are in active phase of cell cycle and threrefore pediatric tumors are more sensitive to chemotherapy and radiotherapy than adult tumors.
Pediatric tumors vs adult tumors similarities
- Accumulation of genetic alterations: the identification of oncogenes (NMYC) and tumor suppressor genes (RB, TP53) in pediatric tumors led to similar findings in adult cancers and vice versa (adult GIST molecular genetic studies lead to definition of pediatric type of GIST)
- Extent of disease/metastatic disease is the one of the most important negative prognostic factors for both – pediatric and adult cancer
- Multimodal treatment: the successful use of intensive multi-agent chemoterapy as curative treatment for pediatric cancer set the example for this approach in adults
Importance of cooperation between pediatric and adult oncology
- experiences of adult oncology: the successful application of targeted therapeutics in adult types of tumors (CML, GIST, renal carcinoma etc) have set the example of this approach for pediatric tumors. The dramatic effect of modern immunotherapy in adult malignant melanoma open the door to use immunotherapy in pediatric oncology
- experiences of pediatric oncology with molecular biology studies, characteristic mutations (oncogen activation, tumor suppressor genes deletion) and translocations lead to molecular genetic phenotypes characteristics for specific types of tumors ( e.g. sarcomas), association of molecular genetic changes with specific morphology and clinical behavior of the cancer leads to more precise risk classification of tumors with direct impact to the cancer management.
- pediatric tumors in adults and adult tumors in children : epithelial types of tumors (carcinomas) are extremely rare in children less than 15 years of the age and also in adolescent age. So called pediatric types of tumors (Ewing sarcoma/PNET, medulloblastoma or rhabdomyosarcoma) may occur also in young adult persons. (Figure 2)
Pediatric oncologist often stands in front of difficult decisions regarding proper and effective treatment strategy for very rare tumors in children and adolescents, because:
- due to their exceedingly rare occurrence do not exist guidelines for optimal treatment
- pediatric oncologist is not familiar with the treatment e.g. adult tumor types
GIT tumors and colorectal carcinoma in children
Definition
Primary malignant gastrointestinal (GIT) tumors in children and adolescents are extremely rare.
Several types of GIT tumor may occur in children and adolescents:
- carcinomas
- neuroendocrine tumors (carcinoid, NETs)
- sarcomas (GIST)
- non-Hodgkin lymphomas
For neuroendocrine tumors (carcinoids) is typical:
- localization at the appendix
- usually benign clinical behavior
- no symptoms of carcinoid syndrome
For pediatric type of GIST is typical:
- gastric localization (often multifocal)
- almost all cases are adolescent girls
- the main symptom is anemic syndrome
For colorectal carcinoma in childhood is typical:
- unfavourable histology type
- advanced stage of disease at diagnosis
- localization common in pericaecal region or colon ascendent
Epidemiology
- GIT tumors account about 1–2% of all pediatric malignancies
- Annual incidence is about 1–1.14 : 1 million for persons less than 20 years of the age
- The higher incidence is in the 2nd decade of the life, in adolescent age (Figure 3)
- GIT tumors are more common in males than in females except neuroendocrine tumors (NETs) and pediatric GIST, which are more frequent in adolescent girls
- The most common GI tumor in pediatric population is neuroendocrine tumor (NET, carcinoid)
Etiology and etiopathogenesis
Etiology and pathogenesis depends on the histologic type of GIT tumor.
For colorectal carcinoma some risk predisposition factors are recognized:
-
Genetic predisposition syndromes (polypous or non-polypous):
- familial adenomatous polyposis
- Lynch syndrome, Turcot syndrome
- Peutz-Jeghers syndrome (Figure 4)
- Inflammatory bowel disease
- Ionising radiation
- Familial occurence (15–20% of patients develop nonpolypous colon carcinoma without defined genetic mutations)
For neuroendocrine tumors (NETs) risk predisposition factors are:
- Familial occurence without defined responsible genetic mutation
-
Genetic predisposition syndromes:
- MEN1 and MEN2 syndromes
- tuberous sclerosis
- neurofibromatosis type I
- Li Fraumeni syndrome
- chronic inflammatory bowel disease
For GIST and sarcomas risk predisposition factors are:
- Sporadic GIST without known risk factors
- Mutation of KIT and PDGFR genes
- Mutation of RB1, SDH genes
-
Specific genetic syndromes:
- Carney and Carney-Stratakis syndrome
- association with neurofibromatosis type 1
For non-Hodgkin lymphomas see chapter non-Hodgkins lymphoma.
Diagnostic procedures
Physical examination: can provide information about palpable mass, bowel movement. Part of clinical examination is digital per rectum examination
Laboratory tests:
- basic hematology exam (CBC, coagulation profile, erytrocyte sedimentation)
- biochemistry (urea, kreatinin, uric acid, liver function tests, ionogram, ALP, AMS, CRP)
- tumor markers: LDH , CEA, Ca19-9 , Ca12-5
-
fecal analysis :
- FOBT (fecal occult blood test)
- FIT (fecal immunochemical test) is more sensitive than FOBT
Endoscopic examination:
- colonoscopy is the „gold standard“ technique for lower GI tract to detect intraluminal colonic lesions, but sensitivity is not 100% (Figure 6 and 7)
- esophago-gastro-duodenoscopy is appropriate diagnostic method for examination of the upper GI tract up to duodenum, but not small intestine
- capsule endocopy may diagnose and evaluate small bowel lesions since capsule passed through small intestine and transmit images.
Imaging studies:
- Plain X-ray: is helpful in a case of suspisious acute abdomen to exclude ileus
- Intestinal ultrasound in experienced hands is able to detect the lesions or polyps of bowel wall, thickeness or defects of the intestinal wall
- CT scan: of the abdomen can detect tumor mass in abdominal cavity, localization, lymph nodes and liver metastases (Figure 8)
- MRI enterography : is helpful to exam parts of the GI tract not visible by endoscopic methods, specially small intestine in younger children.
Histopathology of tumor tissue: endoscopic biopsy of any suspicious lesion or polyp is mandatory to show grade of dysplastic changes specially for patients with genetic syndromes. For patients diagnosed with GI tumor accidentaly is histopathology essential for further treatment and management.
Molecular biology study is mandatory in modern oncology for exact diagnosis, risk stratification, confirmation of subtype of tumor, risk stratification and decision regarding appropriate therapy.
- colon carcinoma: RAS (NRAS,KRAS), BRAF gene
- somatostatin receptors SST1-5, further studies depend on localization of the tumor
- for definition of pediatric GIST is mandatory to evaluate KIT, PDGFRA, SDHA,SDHB,SDHC (Figure 9)
Differential diagnosis
The most important is differential diagnosis of blood in the stool (haematochesis). Causes could be different than in an adult age:
-
Local causes:
- inflammation: inflammatory bowel disease, morbus Crohn, ulcerous colitis, allergic colitis
- infection : bacterial, viral, mycotic, parasitary
- Meckel diverticle
- hemorrhoids, fissura ani
- invagination
- intestinal polyps
- tumor
- Congenital malformations: bowel malrotation, duodenal duplication, intestinal diverticle
- Vascular causes: intestinal angiodysplasia
- Haematologic causes: trombocytopenia, Schonlein-Henoch purpura, thrombosis of mesenteric veins, coagulopathies
- Others: sexual abuse of the child, foreign body in GI tract, drugs (aspirin, nonsteroid anti-inflammatory drugs)
Therapy
Treatment strategy of GI tract tumors is based on the experiences of adult oncology and depends on tumor type, extent of disease and currently also on molecular biology studies. There is still a lack of pediatric studies for GI tumors (Figure 10).
Surgery: the main goal for the surgery is to safely and completely remove the primary tumor. Radical surgical removal of the tumor is mandatory for survival.
For NETs, in children usually in appendiceal localization, surgery (appendectomy) is curative without any further anticancer treatment.
For tumors > 2cm, more aggressive histopathology findings localised at the base of appendix right hemicolectomy is indicated.
For GIST surgery was the only therapeutic modality for many years. In case of localised completely resected GIST surgery is also curative. Type of surgery depends on localization of GIST and number of lesions.
For colon carcinoma radical surgery (hemicolectomy) is one of the most important factors influencing prognosis.
Chemotherapy: for pediatric GI tumors systemic chemotherapy is indicated usually for colon carcinoma in adjuvant setting. Treatment schedules and guidelines are based on experiences and recommendations of adult oncology.
For pediatric GIST and NETs chemotherapy is largely ineffective.
Biology targeted therapy: historically started in the year 2000 with using imatinib mesylate (glivec) in the treatment of GIST in adult population. For pediatric type of GIST with different molecular genetic signature effect of glivec is not well documented, therefore multikinase inhibitors are used as the first line therapy (sunitinib).
For NETs in pediatric age usualy targeted therapy is not indicated due to favorable localization and complete surgical resection. For metastatic or widespread NETs somatostatin analogs are indicated, mTOR inhibitors are used for advanced pancreatic or lung NETs.
For colon cancer biology targeted therapy indication is based on extent of disease and molecular genetic studies. For advanced stage addition of bevacizumab (anti VEGF) or cetuximab (anti EGFR) to chemotherapy significantly improved outcome.
Prognosis and outcome
Prognosis of pediatric GI tumors depends on type of tumor, localization, extent of disease and underlying genetic condition.
Outcome of colorectal carcinoma is worse than in adults and depends on:
- the age of the patient and underlying genetic condition
- extent of the disease (clinical stage)
- molecular genetic backgroud (RAS mutation,BRAF mutation)
Outcome of neuroendocrine tumor depends on:
- localization of the tumor (appendix vs small intestine) and possibility of radical surgery
- grade and size of the tumor
- extent of disease
Outcome of pediatric GIST depends on:
- extent of disease and possibility of radical surgery
- molecular genetic studies ( mutation of KIT,PDGFR, SDH genes)
5-year overall survival:
colorectal carcinoma 40–60%
neuroendocrine tumor (carcinoid) > 98%
pediatric GIST 65–75% (Figure 11)
Author: Viera Bajčiová, MD, PhD
Breast tumors in childhood and adolescents
Definition
Breast cancer is extremly rare in children and adolescents. Less than 0.2% of all breast cancer in population is diagnosed in girls younger than 20 years. Breast masses or lesions may occur in girls as well as in boys.
Breast lesions in boys are extremely rare. Main problems of the breast in boys are:
- gynecomastia
- accessory breast/nipple (Figure 12)
- inflammatory lesions (borreliosis, absces)
- benign tumors (lipoma,fibroma)
- malignant tumors
Spectrum and biology behavior of breast masses in adolescent girls differ from the breast masses in adult women (Figure 13).
The most common breast masses in pediatric population are tumor-like lesions and juvenile fibroadenoma (Figure 14).
Breast carcinoma is exceedingly rare in young population, but diagnosed in adolescents is more aggressive and usually more advanced stage than in older age. Adolescent breast carcinoma is characterised:
- frequently invasive ductal type
- triple negative (hormonal and HER2receptors are negative)
- high proliferation activity
- positive lymphovascular invasion
Pediatric ICCC classification due to rarity of breast cancer do not have specific code for this diagnosis and therefore code XI (other or unspecified carcinoma) is used in case of this rare diagnosis
Epidemiology
Incidence of juvenile fibroadenoma is 5 : 1 million, characteristic age 14–19 years.
Incidence of breast carcinoma:
- 15–19 years 0.8 : 1 million
- 20–24 years 1.21 : 1 million
Annual incidence of breast carcinoma in young popualtion is stable (Figure 15).
Incidence of sarcoma of the breast is < 1 : 1 million, characteristic age > 16 years.
Breast masses are extremely rare in children under 10 years of the age.
Etiology and etiopathogenesis
Based on rarity of breast tumors in young generation is only very limited number of epidemiology studies focused on pediatric and adolescent age.
Fibroadenoma occurence is sporadic in most of the cases. Some risk factors are under the debate:
- hormonal status may play a role (contraceptives, pubertal hormonal changes)
- genetic syndromes: Beckwith-Wiedemann syndrome, tuberous sclerosis
- hemihypertrophy
Carcinomas of the breast are associated with some risk factors:
- familial occurence without known causal mutation
-
hormonal factors:
- early menarche (<11 years of the age)
- exogenous hormones (contraceptives)
- benign breast disorders (cystic dysplasia with cellular atypia)
- genetic predisposition:
- mutation BRCA1/BRCA 2 tumor suppressor genes
- Cowden syndrome (mutation of PTEN gene)
- Li Fraumeni syndrome ( mutation of TP53 gene)
- Peutz-Jeghers syndrome ( mutation of STK11 gene)
- environmental factors: ionising irradiation ( CAVE – risk of secondary breast carcinoma for Hodgkin lymphoma survivors )
- life style factors (obesity, smoking, alcohol, stress, low physical activity)
Sarcoma of the breast – genetic changes (mutations) depend on type of sarcoma
Clinical presentation and symptoms
Local symptoms:
- palpable lump or thickening in the breast or in axilla,
- asymmetry of the breast
- nipple changes - tenderness, nipple turned slightly inward or inverted , any nipple discharge
- a change of the skin texture ( orange peels texture)
Systemic symptoms: general weakness, anorexia ,weight loss, bone pain or other non specific complains
Diagnostic procedures
Personal and family history: duration of the symptoms,, puberta and medical history, menarche. Previous malignancy and its treatment. Important is information of family breast and ovarian cancer
Physical examination may be difficult due to a broad variability of the glandular size during childhood and puberty. Location, size, consistency, mobility, pain of lump, skin changes and nipple appearance have to be evaluated. The clinical examination must always include the axillary lymp nodes
Very important is education of adolescent girls about technique of breast self-examination. (Figure 16).
Laboratory test:
- hematology – blood tests (CBC, coagulation profile)
- biochemistry – liver enzymes, ALP,LDH, urea, creatinin, uric acid
- tumor markers – Ca15-3, ferritin, CEA
Imaging studies):
- Ultrasound: of adolescent breast is sometimes difficult to asess due to high density of juvenile breast. Is useful to recognised cystic vs solid lesion, size, localization, vascularisation, density and lymph nodes enlargement and character. For diagnosis of juvenile fibroadenoma the UZ picture is typical (Figure 17).
- Mammography: is usually not used for children and adolescents with breast mass due to high radiation exposition and its diagnostic inaccuracy.
- MRI of the juvenile breast: is important for detailed investigation of breast lesions, vascularisation, character. It is indicated in case of suspicious malignancy. MRI of juvenilie fibroadenoma show lobulated well circumscribed mass with internal non engancing septa.
- Staging CT scan and bone scan: to confirm/or exclude metastatic spread to the lung and bones
Histopathology:
- Type of tumor: is evaluated with fine needle aspiration biopsy, if lesion is suspected from malignancy. Carcinoma in situ is exceedingly rare in adolescents. The most common type of carcinoma is secretory carcinoma. About 45% of tumors in youngs are phylloid tumors with indolent clinical course. Invasive ductal carcinoma is aggressive tumor with unfavourable prognosis.
- Hormone receptors status ( estrogen, progesteron receptors and HER2 receptors) (Figure 18 and 19)
- Molecular genetic studies: in familial occurrence of breast cancer or ovarian cancer genetic counseling two tumor-suppressor genes BRCA1 (Breast cancer 1) and BRCA2 (Breast Cancer 2) is used (Figure 20). Products of both BRCA genes participate in the regulation of cell cycle progression and DNA repair. Inherited mutations of BRCA1 is found in families with familial incidence of breast and ovarian cancer. BRCA2 germline mutation is associated with the incidence of breast cancer of women and men with tumorigenesis of prostate, pancreas, and Fanconi anemias.
Differential diagnosis
Differential diagnosis of the most frequent juvenile fibroadenoma:
-
Benign lesions:
- virginal hypertrophy of the breast
- premature telarche
- fibrocystic disease, ductal cysts of the breast
- intraductal papilloma
- haemangioma/lymfangioma
-
Malignant lesions:
- malignant phylloid tumor (cystosarcoma phylloides)
- sarcoma ( rhabdomyosarcoma, angiosarcoma)
- metastases to the breast (alveolar rhabdomyosarcoma, neuroblastoma, Ewing sarcoma/PNET, malignant melanoma etc.)
- carcinoma
Therapy
Due to rarity of breast masses in adolescent girls is very diffucult to establish treatment guidelines for this age specific group of patients. No age specific treatment guidelines exist yet and therefore guidelines followed recommendations of adult oncology.
Fibroadenoma
Strategy watch and wait is recommended for fibroadenomas with following feratures:
- size < 5 cm or volume < 500ml
- no clinical symptoms, no pain or other problems
- no documented growth of the lesion
- characteristic UZ picture, no atypia and no changes in time
Surgery for fibroadenoma is indicated if one or more features mentioned above is not fulfilled. Goal of the surgery is radical resection of the lesion, but with sparing of breast parenchyma – lumpectomy and reconstructive techniques. Ablative surgery (mastectomy) is not indicated as well as resection of regional lymph nodes.
Carcinoma
Invasive ductal form in adolescent age has an extremly aggressive clinical behavior and therefore very aggressive treatment is required in form of radical mastectomy, regional lymphadenectomy and aggressive chemotherapy. Breast carcinoma in young population is frequently so called “triple negative” cancer (estrogen, progesteron and HER2 receptors are negative), therefore chemotherapy is treatment of choice, usually based on anthracyclines and taxanes. Hormonal therapy and targeted biology therapy is ineffective.
Clinical studies phase 1 and 2 with new treatment modality (immunotherapy with antiPD-1 monoclonal antibody) are on the way, but only for adult patients. No pediatric age specific clinical study exists.
Metastases of other tumors to the breast
Usually is a part of widespread tumors (soft tissue sarcomas), treatment is based on guidelines for primary tumor or palliative treatment.
Prognosis and outcome
Prognosis of the breast tumor depends on:
- type and grade of the tumor
- age at diagnosis is important and independent prognostic factor
- extent of disease (clinical stage)
- hormonal receptors expression
- genetic mutations (BRCA1,2, TP53)
5-year overall survival for carcinoma generally is about 65–80%. Almost all patients with juvenile fibroadenoma survive 5 years.
5-year overall survival for primary breast sarcomas is nearly to 80%, for metastatic disease prognosis is dismal (Figure 21).
Author: Viera Bajčiová, MD,PhD
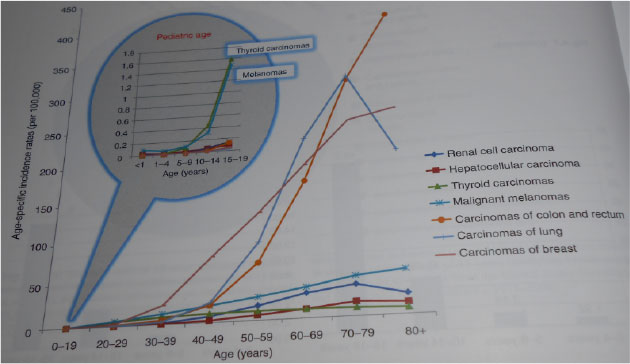 Figure 1:Age specific incidence of adult types of tumors in childhood (SEER 1973–2006) |
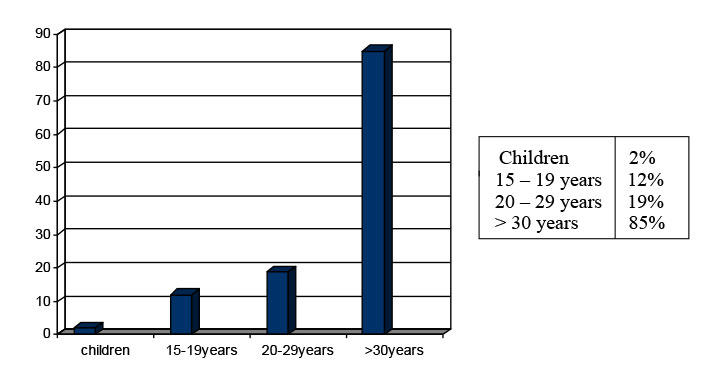 Figure 2: Age distribution of epithelial cancer (carcinoma) |
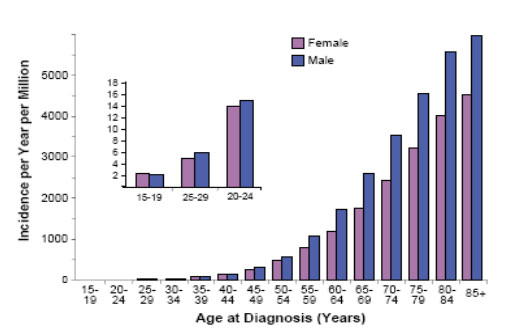 Figure 3: Incidence of colorectal carcinoma (SEER, 1975–2000) |
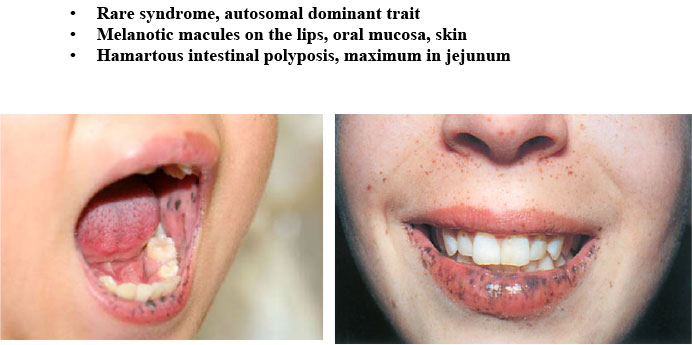 Figure 4: Peutz-Jeghers syndrome |
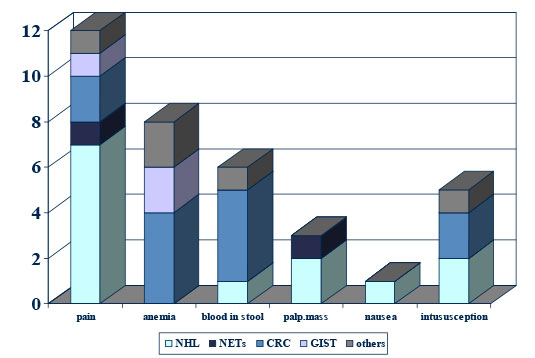 Figure 5: Clinical symptoms of GI tumors in childhood |
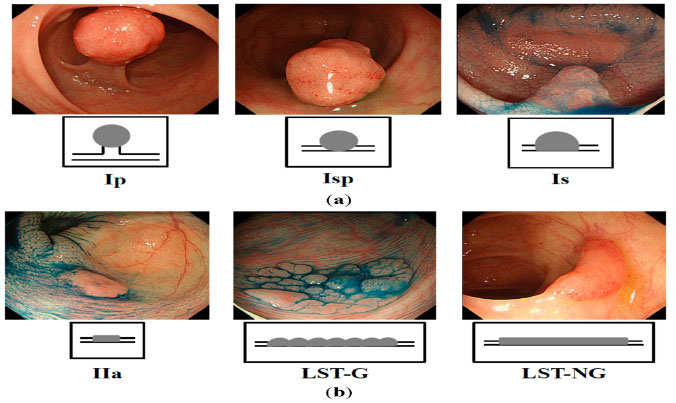 Figure 6: Endoscopic types of colon polyps ( Nakagawa Y, Int J Mol Sci 2015) |
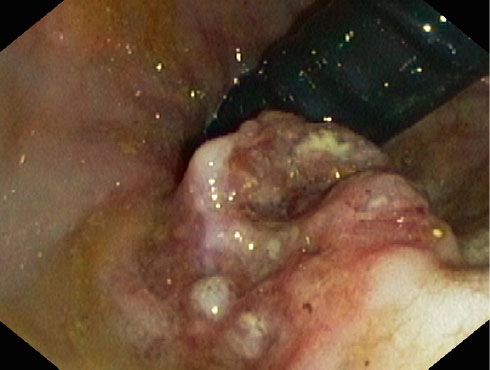 Figure 7: Endoscopy of adenocarcinoma of the colon |
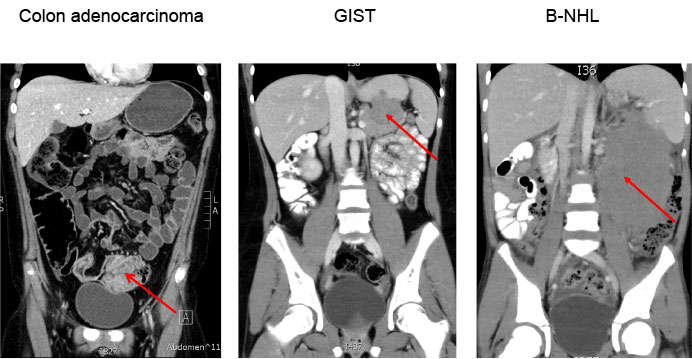 Figure 8: Initial abdominal CT scan |
 Figure 9: Differences between pediatric and adult GIST |
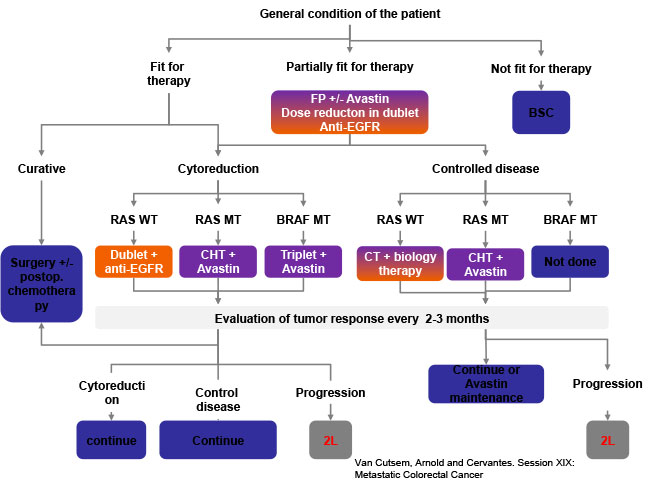 Figure 10: ESMO 2015 consensus on treatment of advanced mCRC |
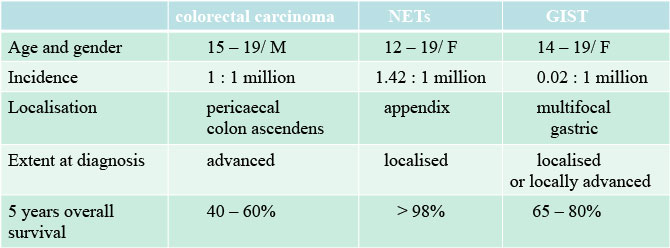 Figure 11: GIT tumor in children and adolescents |
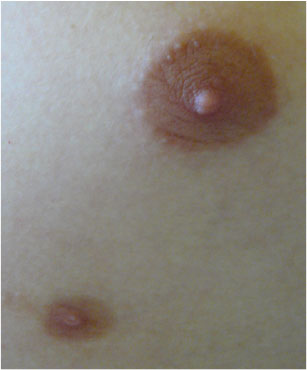 Figure 12: Accessory nipple (KDO FN Brno) |
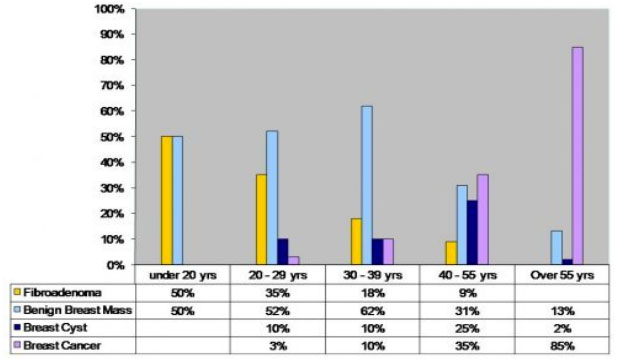 Figure 13: Spectrum of breast lesions according to age |
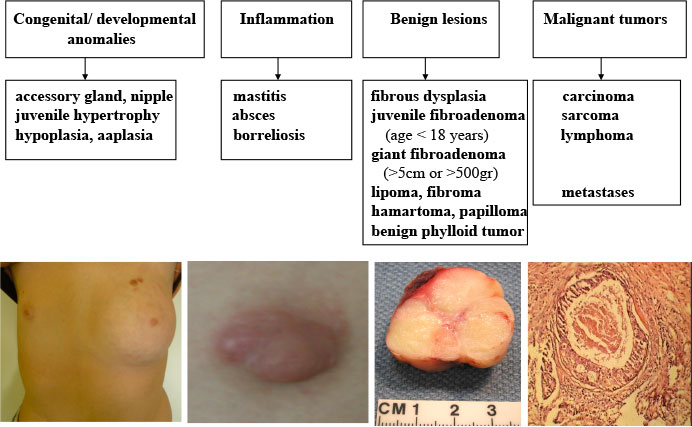 Figure 14: The most common breast pathology in adolescent girls |
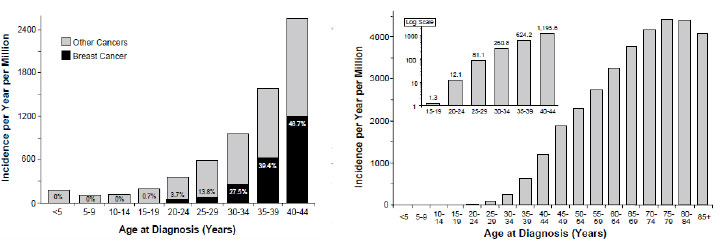 Figure 15: Incidence of breast cancer in females according to age (Bleyer WA, 2007) |
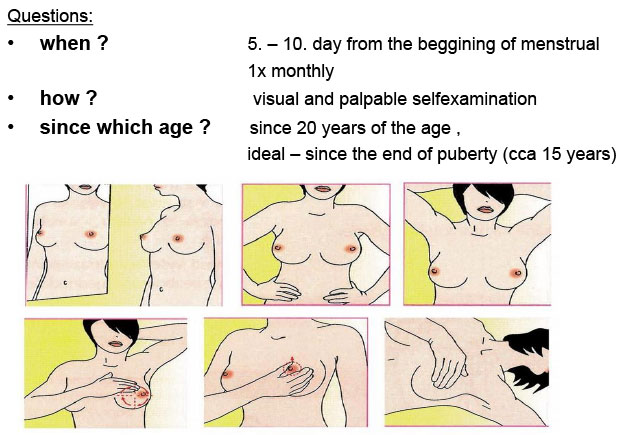 Figure 16: Breast self-examination |
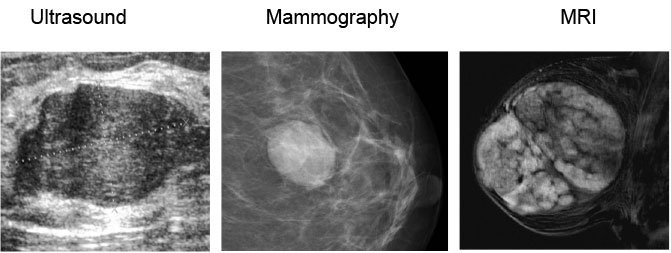 Figure 17: Imaging studies for juvenile fibroadenoma |
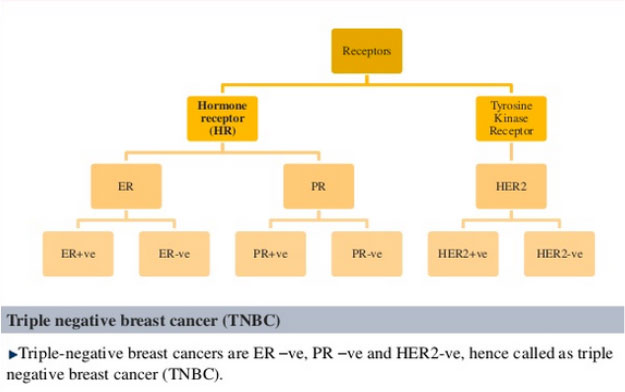 Figure 18: Receptor overexpression specific for breast cancer (Jadhav A, 2013) |
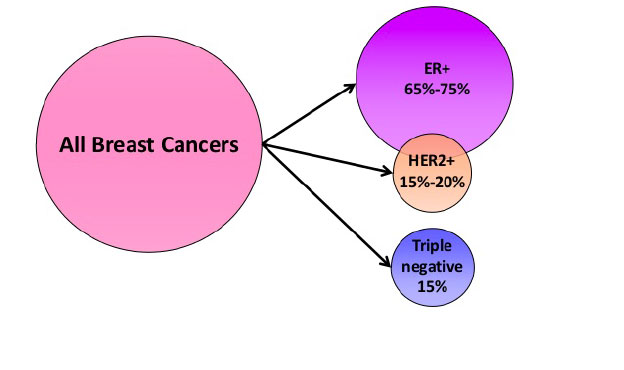 Figure 19: Clinical breast cancer subsets defined by receptors overexpression |
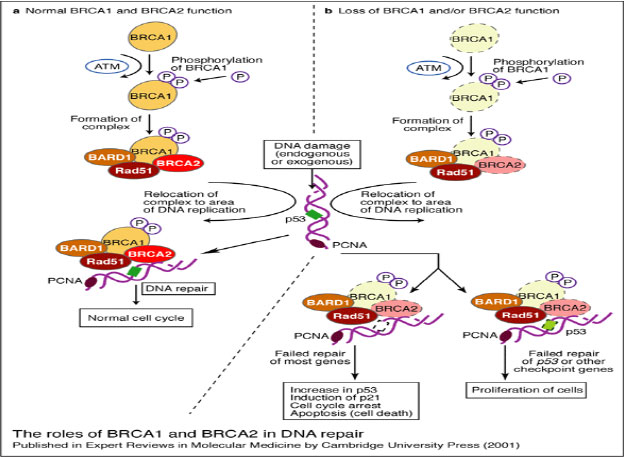 Figure 20: Tumor supressor genes BRCA1, BRCA2 (Exp Rev Mol Med 2001) |
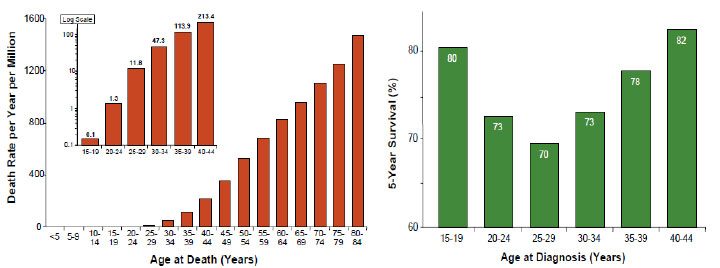 Figure 21: Mortality rate and 5 years overall survival for breast cancer according to age (Bleyer AW, 2007) |
|||