E-learning 2. Analýza dat 2.2. Analýza vysokopokryvných genomických dat 2.2.1. DNA mikročipy 2.2.1.8. Analýza CGH čipů
Věří se, že současné hledání aberovaných oblastí napříč vzorky ze stejné skupiny může usnadnit hledání specifické skupiny aberací.
Jeden z prvních přístupů tohoto druhu byla navržen v [42]. Tento přístup nejprve aplikuje GLAD algoritmus [20] k detekování oblastí a přiřadí jim jednen ze stavu - normál, deleci nebo amplifikaci. Výsledky z této analýzy jsou řazeny do maticového tvaru, kde sloupce reprezentují sondy a řádky představují vzorky, 0 znázorňuje nepřítomnost aberace a 1 buď deleci nebo amplifikaci nacházející se v pozici příslušného vzorku. Delece a amplifikace jsou zpracovány v samostatných maticích. Tyto matice jsou dále využívány pro detekci opakujících se oblastí v rámci skupin. Opakující se oblast je definována jako sekvence změněných sond společný souboru CGH profilů a minimální opakující se oblast (MAR) jako opakující se oblast, která neobsahuje menší opakující se (MARs). K hledání MARs byly představeny dva algoritmy. Ty jsou založeny na hledání bloků sond s podobnými stavy získanými z GLAD napříč všemi vzorky. První algoritmus, MAR, efektivně vypočítá všechny minimální opakující se změny oblastí ze souboru diskretizovaných profilů. Druhý algoritmus, CMAR, umožňuje začlenit předchozí biologické relevantní informace, jako je minimální četnost daných oblastí změn v datovém souboru nebo počet pozorování definující hranice změněné oblasti a tím minimalizovat možná příliš velký počet MARs nalezených prvním algoritmem. Podobný přístup byl představen v [11]. Vstupní data pro tento algoritmus jsou ve stejné maticové formě jako v [42]. Poté se používá statistické testování k nalezení odpovídajících aberací napříč vzorky. Nulová hypotéza je, že u pozorovaných segmentů aberací je stejně pravděpodobné, že se vyskytnou v jakékoli zvažované části genomu. Nulová distribuce je získána prostřednictvím náhodného znovu uspořádání segmentů každého vzorku bez vrácení.
Nicméně, tyto přístupy k identifikaci opakujících se oblastí jsou spíše přirozeně post-zpracovávající než části segmentační procedury. Společná segmentace může předejít odfiltrování důležitých signálů vyskytujících se v původních datech. Jeden z prvních relevantních přístupů provádějících společnou segmentaci profilů přes vzorky byl navržen v [27], který poskytuje analýzu segmentových stavů. Tato metoda je založena na předpokladu, že v rámci nulové hypotézy nejsou objeveny v datech žádné změny. Statistika se rovná
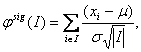
kde I je segment a řídí se normálním rozdělením s m=0 a s2 =1 pro každé I. Použitím této statistiky je stanovena statistická významnost pro každý segment. Všechny intervaly, jejichž statistiky překročí předem definovanou hranici, jsou považovány za domněle aberované. Tento jedno-vzorkový případ může být rozšířen na více vzorků, umožňující dva způsoby analýzy: fixní mód, kde se provádí hledání aberací přítomných ve všech vzorcích, a mód objevování třídy, kde je prováděno hledání aberací přítomných v podsouborech vzorků. Engler et al. [13] navrhli tří-stavový smíšený Gaussův HMM model s parametry, který je považovaný za běžný nejen napříč chromozomy, ale taky vzorky. Také představili prostorovou závislost dat a navrhli provedení klasifikace na základě posteriorních pravděpodobnostech stavů. Další společný segmentační přístup byl prezentován v [47]. Tento přístup rozšiřuje jedno-vzorkový HMM model na vícevzorkový případ. Jsou navrženy tři metody: promítnutý pravděpodobnostní HMM, promítnutý pravděpodobnostní HMM s vyrovnávací pamětí a hierarchický HMM. Shah et al. [47] oznámili vynikající výkon posledně zmíněného modelu. Další zlepšení vyhlazovacích přístupů pro analýzu dat z CGH čipů lze získat zavedením dvojité važené konečné metody díky [19]. Ta může být rozšířena na situaci více vzorků zachycujících skupinu efektů společným odhadem parametrů.
Konečně nová metoda navržena [37], kde je efektivní segmentační přístup [35] zobecněn pro případ více profilů. Jejich přístup využívá smíšený lineární model s příslušnými bodovými zlomy. Parametry modelu jsou odhadovány pomocí metody maximální věrohodnosti, získané EM algoritmen kombinovaným s dynamickým programováním (DP).