E-learning 2. Analýza dat 2.2. Analýza vysokopokryvných genomických dat 2.2.1. DNA mikročipy 2.2.1.8. Analýza CGH čipů
Tzv. segmentační metody se pokouší rozdělit data do dobře rozlišitelných segmentů s různým průměrem log2 podílů. U klonů v rámci odhadovaného segmentu se předpokládá, že sdílejí stejný počet kopií, a hranice segmentů jsou často označovány jako zlomové body (nebo hrany). Různé přístupy odhadů jsou převzaty k lokalizaci segmentů na genomu a optimálnímu počtu segmentů.
Model založený na segmentačních technikách upravuje model na data a zkouší identifikovat zlomové body způsobem, který maximalizuje pravděpodobnost rozdělení pravděpodobnostní funkce.. Mohou se lišit v objektivní funkci a použitém optimalizačním kritériu. Většina z těchto metod modeluje data podle Gaussovského procesu a provádí analýzu každého chromozomu zvlášť.
Princip modelu založeného na segmentačních technikách
Pro větší konkrétnost, nechť ![]() představuje naměřené log2 podíly pro daný chromozom v pozici 1…N, hlavním cílem je rozdělení těchto hodnot do segmentů
představuje naměřené log2 podíly pro daný chromozom v pozici 1…N, hlavním cílem je rozdělení těchto hodnot do segmentů ![]()
![]() …
… ![]() tak, aby počet kopií klonů ve stejném segmentu byl stejný. Indexy
tak, aby počet kopií klonů ve stejném segmentu byl stejný. Indexy ![]() kde y0=1 a yk=N, představují pozici bodových zlomů (nebo jednoho zlomu). Předpokládá se, že pro všechna x ze stejného segmentu jsou parametry Gaussova rozdělení konstantí:
kde y0=1 a yk=N, představují pozici bodových zlomů (nebo jednoho zlomu). Předpokládá se, že pro všechna x ze stejného segmentu jsou parametry Gaussova rozdělení konstantí: ![]()
![]()
Vzhledem k tomu, že jsou klony považovány za nezávislé, může být pravděpodobnostní funkce definována jako:
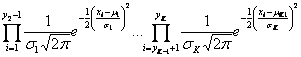
Maximalizace této funkce podle ![]() vede k odhadu optimálního rozdělení dat. To je ekvivalentní k minimalizování logaritmu negativní pravděpodobnostní funkce, jejíž výsledky v součtu dílčích log-pravděpodobností jsou:
vede k odhadu optimálního rozdělení dat. To je ekvivalentní k minimalizování logaritmu negativní pravděpodobnostní funkce, jejíž výsledky v součtu dílčích log-pravděpodobností jsou:
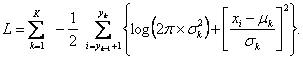
Hledání optimálního rozdělení dat (optimalizace), kde je K známé, lze dosáhnout pomocí různých algoritmů.
Dalším úkolem je zvolit vhodné K. Je zřejmé, že čím vyšší je počet zlomových bodů (a také segmentů), tím lze dosáhnout vyšší pravděpodobnosti. Penalizace zabraňuje nadhodnocení počtu zlomových bodů. Pokud označíme LK hodnotou log-pravděpodobnosti nejlepšího rozdělení dat v K segmentech, pak optimální K lze odhadnout minimalizací následující rovnice:
![]()
kde pen(K) je funkce parametrů modelu takového, že funkce vzrůstá s rostoucím počtem parametrů a b je penalizační konstanta. Tabulka 1 ukazuje různé penalizace použité v analýze CGH čipů (podle [35]).
Jeden z prvních přístupů modelu založeného na segmentaci byl navržen Jongem et al. [21]. Genetický lokální vyhledávací algoritmus je aplikován tak, že najde nejpravděpodobnější rozdělení dat minimalizací penalizovaného maximálního pravděpodobnostního kritéria. Picard et al. [35] předpokládají homogenní rozptyl a vhodný model pomocí algoritmu dynamického programování. Poukazují na to, že konstantní penalizační koeficient pravděpodobně způsobuje „nadsegmentaci“ v modelu a doporučili adaptivní penalizační kritérium původně navržené v [25].
Tabulka 1. Různé penalizační funkce použité při analýze CGH čipů
|
kritérium |
b |
pen(K) |
|
AIC |
1 |
2K |
|
BIC |
log(n) |
2K |
|
Jong et al. [21] |
10/3 |
3K-1 |
|
Lavielle [25] |
adaptativní |
2K |
Rekurzivní segmentační technika, tzv. kruhová binární segmentace (CBS) je trošku odlišný a poněkud jednoduchý přístup představený v [33]. Hlavním principem je výpočet určité Z-statistiky která je porovnána s hodnotou teoretického rozdělení. Tato statistika se vypočte následovně: Nechť ![]() je pořadí log2 podílů pozorovaných na chromozomu a nechť
je pořadí log2 podílů pozorovaných na chromozomu a nechť ![]() jsou parciální součty. Za předpokladu, že známe rozptyl s2 pro všechna xi, Z-statistika je definována jako
jsou parciální součty. Za předpokladu, že známe rozptyl s2 pro všechna xi, Z-statistika je definována jako![]() , kde Zij je dvouvýběrová t-statistika pro porovnání průměru pozorování s indexem od i+1 do j, k průměru zbylého pozorování:
, kde Zij je dvouvýběrová t-statistika pro porovnání průměru pozorování s indexem od i+1 do j, k průměru zbylého pozorování:
![]()
Podle nulové hypotézy o žádné změně, mají všechny Zij normální rozdělení s nulovou střední hodnotou a společným rozptylem s2. Zlomový bod je detekován, když Z překročí a-tý kvantil normálního rozdělení (nebo permutaci na základě referenční distribuce). Tento proces se opakuje rekurzivně v zjištěných segmentech, dokud není nalezena žádná změna v žádném z kontrolovaných segmentů. Výpočetní zátěž jejich algoritmu byla nedávno snížena na lineární dobu v [52], kde je představen hybridní přístup pro výpočet p-hodnoty testové statistiky a nové ukončovací pravidlo.
Další přístup byl navržen na základě třístupňového zlomového detekčního algoritmu v [30]. Nejprve je pro odhad umístění bodových zlomů použit hraniční detekční filtr obsahující sekvence filtrovacích kroků. Poté je použit EM-algoritmus poskytující jemné úpravy odhadů bodových zlomů. Nakonec jsou prováděny různé statistické testy k posouzení významu zjištěných segmentů.
Žádný z těchto algoritmů neposuzuje detekované oblasti. Výstup segmentačních metod se obvykle skládá ze zlomových bodů a průměru segmentů. Segmentační metody tak potřebují další analýzu, jejíž cílem je zařazení odhadovaných segmentů do biologicky významných kategorií (delece, normál, amplifikace).
GLAD
Hupé et al. [20] byli jedni z prvních, kteří se tímto aspektem zabývali. V jejich přístupu nazvaném GLAD, lokální Gaussovský regresní model pro každé pozorování xi předpokládá:
![]()
Parametr q je lokálně odhadován pro každou pozici pomocí adaptivní vážené vyhlazovací (AWS) procedury [40]. Když je pro každý klon získán parametr odhadu, jsou zlomové body stanoveny v pozicích, kde se qi liší o více než e=10-2 od qi+1. Optimální počet zlomových bodů je pak odhadován pomocí penalizovaného pravděpodobnostního kritéria, které používá „trojkubickou“ kernelovu funkci. Po zabudování modelu se pro posuzování oblasti použije dvoukroková (chromozomální a genomové úrovně) shlukovací procedura. Princip je následující: na chromozomální úrovni jsou data shlukována prostřednictvím hierarchického shlukování se středovými kritérii s přihlédnutím k počtu pozic v každém segmentu. Pak je takto získaný dendrogram postupně seříznut kvůli získání souboru shluků. Optimální počet shluků je vybrán opět pomocí penalizačního pravděpodobnostního kritéria. Tento proces je opakován na genomové úrovni se shluky vybranými v chromozomové úrovni. GLAD také věnuje zvláštní pozornost identifikaci odlehlých hodnot a jejich odstranění v každém kroku analýzy (jak v detekci zlomových bodů, tak ve shlukování).
Picard et al. [36] nedávno vylepšili svou segmentační metodu [35] segmentačně/shlukovacím modelem. Algoritmus dynamického programování předpokládající maximalizaci (DP-EM) je alternativně použit k odhadu souřadnic zlomových bodů a parametrů smíšeného Gaussovského modelu. Také je navržena heuristická technika pro jednotné vybrání počtu segmentů a počtu shluků, založená na penalizačním pravděpodobnostním kritériu. Další segmentační metodu přihlížející ke klasifikaci navrhli Broët & Richardson [6]. Jejich metoda využívá Bayesův prostorově strukturovaný smíšený model a poskytuje posteriorní pravděpodobnosti patřící k místu k pro každý klon i. Podobně jako v předchozích dvou metodách, aplikuje tato technika klasifikační proceduru, ale také je u ní navržen odhad FDR. Obě klasifikační procedury a odhad FDR jsou založeny na posteriorních pravděpodobnostech. V práci van de Wiel et al. [51] je kombinováno několik koncepcí již navržených metod. Nejprve CBD [33] provádí segmentaci dat, poté jak je navrhováno v [13] jsou použity náhodné efekty a nakonec smíšený model pro segmentaci výsledků pomáhá získat nejvíce pravděpodobnostní klasifikaci segmentů. Aby bylo možné sledovat biologické pozadí dat, uvažuje algoritmus šesti-stavový model, ale konečná klasifikace, která je založena na odvození pravidel z posteriorních pravděpodobností, je nastavena na tři nebo čtyři stavy.
K řešení problému klasifikace detekovaných segmentů přispěli Willenbrock & Fridlyand [55]. Navrhli metodu MergeLevels, která provádí posteriorní analýzu dat ze segmentačních algoritmů. Metoda slučuje odhadované segmenty podle jejich průměrných log2 podílových hodnot kombinací testovací hypotézy (pro širší segmenty) s prahováním (pro segmenty s méně než 3 klony). Dva segmenty jsou sloučeny v případě, že rozdělení log2 podílů klonů příslušných k těmto segmentům se výrazně neliší, nebo když předpokládané hladiny hodnot jsou blíže, než je stanovená hranice. Tento postup se opakuje kvůli zvýšení prahové hodnoty a pro každou hranici se použije Ansari-Bradleyův dvouvýběrový test [4] k určení, zda se rozdělení současného rezidua (aktuální sloučené hodnoty mínus pozorované log2 podíly) výrazně liší od původních reziduí (původní segmentační hodnoty mínus pozorované log2 podíly). Optimální hranice je vybrána jako největší hranice, pro kterou je Ansari-Bradley p-hodnota větší než 0,05 (kde se dva typy reziduí významně neliší).
Segmentační metoda zaměřená na analýzu dat získaných ze SNP čipů byla vyvinuta teprve nedávno [56]. Pro identifikaci zlomových bodů je použita dvoukroková metoda. První krok se vyznačuje vysokou citlivostí na jednu stranu a nízkou na stranu druhou, a jeho cílem je identifikovat co nejvíce pravých zlomových bodů. Tento krok je založen na loess vyhlazování a zlomový bod je identifikován ve středu každé monotónní části vrcholu ve vyhlazených datech, kde se velikost této části nachází nad předem definovanou hranicí. V druhém kroku se k odstranění falešných zlomových bodů používá statistické testování průměrných log2 podílů dvou segmentů kolem každého bodu zlomu. Toto testování používá „forward-backward“ algoritmus, u kterého je možné již odstraněný bod zlomu vrátit, pokud se jedná o nejvýznamnější ze všech odstraněných bodů zlomu a pokud je významnější než ten právě odstraňovaný. Autoři porovnávají svou techniku s několika dalšími metodami a referují vysokou citlivost a vysokou specificitu v detekci malých změn počtu kopií. Zdá se, že tato metoda je dobře výkonná, i když jsou data velmi zanesená šumem.
MSMAD
Další segmentační přístup s vysokou účinností, speciálně navržený pro velmi hlučné data získané z CGH čipů, byl navržen Budínskou et al. [7]. Tato metoda zvaná MSMAD je poměrně jednoduchá a výhradně neparametrická, přijímá koncept střední absolutní odchylky pro detekci zlomových bodů a zahrnuje v předzpracování střední vyhlazování [12]. Ačkoli je metoda vysoce citlivá, měla by pro konečné zpracování použít MergeLevels proceduru, kvůli snížení počtu falešných bodů zlomu a zlepšení konečného výsledku hodnocených oblastí. Jednou z hlavních výhod této metody je, že specifikační parametr v MSMAD je více intuitivní a srozumitelnější, než v mnoha jiných algoritmech, což by mohli ocenit uživatelé nezabývající se bioinformatikou. Ještě jednu významnou výhodu tento algoritmus má - je vhodný pro velmi velké soubory dat (vyplývající z nových vysokopokryvných CGH čipů), protože je výpočetně efektivní a má malé nároky na paměť.