Special section Bone tumors Ewing sarcoma / PNET family tumors
Definition
The Ewing family of tumors is group of cancers of neuroectodermal origin that starts in the bones or nearby soft tissues that share some common biology features.
The main types of Ewing tumors are:
- Ewing sarcoma of the bone – it is the most common tumor in this family
- Extraosseous Ewing tumor (EOS) start in soft tissues around the bone (also known as extrasceletal Ewing sarcoma) and can occurs almost anywhere
- Peripheral primitive neuroectodermal tumor (PPNET) starts in bones or soft tissues; when starts in the chest wall, is also known as Askin tumor
Ewing sarcoma of the bone was identified in 1921 by James Ewing and accounts 3% of all pediatric malignant tumors
The most common localization of Ewing sarcoma (Figure 1):
- diaphysis of the long bones (53%)
- pelvis (25%)
- chest wall (18%)
- spine (8%)
- head and neck (4%)
In contrast, osteosarcoma usually occurs at the end (epiphysis) of the long bones.
Epidemiology
- Ewing sarcoma accounts for 10–15% of all primary malignant bone tumors and it is the 2nd most common bone cancer in teenagers
- Annual incidence is estimated 1.5 – 3 : 1 million (Figure 2)
-
The overall male: female ratio:
- age < 10 years 1.3 : 1
- age > 10 years 1.6 : 1
- There is higher geographic and race heterogeneity than in osteosarcoma, Ewing sarcoma is less frequent in black children and Asian population,
- Bimodal age curve is noticed in Ewing sarcoma, it occurs commonly in 2nd decade, but also can occurs in small children about 5 years of life
Etiology and etiopathogenesis
Almost all Ewing sarcoma/PNET tumors are sporadic without known risk factors.
- Studies of Ewing sarcoma have not found links to radiation, chemicals, diet or any other environmental exposures
- Hereditary genetic changes are not an important risk factor for Ewing/PNET tumors. Although the genetic changes that cause most Ewing tumors (EWS/FLI 1) are not inherited
- Familial occurrence is rare
- No hereditary syndromes predispose to Ewing sarcoma
Clinical presentation and symptoms
Local symptoms depends on location and size of the tumor:
- Local pain – night pain is typical, „on-off“ character
- Lump or swelling of the affected area, rapid increase in size. Lump is often soft and feels warm (Figure 3)
- Impairment of function, changes of movement stereotype
- Pathologic fracture (Figure 4)
- Compression and obstruction of surrounding organs (bowels, spine, ureter)
- Duration of the symptoms is several months (4–6 months)
Systemic symptoms:
- Weakness, anorexia, weight loss
- Fever not resolving to antibiotics
- Night sweat
- Paraneoplastic laboratory: high CRP, ESR, LDH
Symptoms related to metastatic disease: 25% patients have metastatic disease at the time of diagnosis. Metastases generally spread through blood stream to the lung, other bones and bone marrow.
- Lung metastases (38%) – cough, tachypnoe, dyspnoe (Figure 5)
- Multiple bone involvement (31%) – pain, systemic symptoms
- Bone marrow (11%) – thrombocytopenia, anemia
Diagnostic procedures
The diagnostic work up includes family and personal history, type and duration of symptoms.
The main goal of diagnostic procedures is an exact description of the primary lesion and a search for metastases (Figure 6).
Physical examination: paying special attention to any areas causing pain or swelling
Laboratory tests:
- usually show elevated ESR (erythrocyte sedimentation rate), some degree of anemia or thrombocytopenia
- elevated marker of inflammation (CRP)
- elevated serum level of LDH is thought to be of prognostic significance (correlate with tumor burden)
- in some patients elevation of serum neuron specific enolase (NSE) possibly related to more neurally differentiated sarcomas
- detection of fusion protein EWS/FLI from peripheral blood and bone marrow
Imaging studies:
-
Plain Y-ray: show lytic or mixed lytic-sclerotic areas localized in diaphysis of the long bones (Figure 7)
- character moth eaten bone destruction
- multi-layered subperiosteal reactions (onion skin) or hair-on-end
- Codman triangle
- the whole bone and nearest joint have to be seen on X-ray
- CT scan: of affected bone as well as CT scan of the chest is a part of initial work up. Bone destruction, intramedullary space and extraosseal involvement, transarticular spread is better seen on CT than on X-ray (Figure 8)
- MRI: detect extends beyond the anticipated area. MRI is crucial before planning surgery to see relationship with neurovascular structures. Also it is mandatory for treatment response evaluation (Figure 9)
- 18 FDG PET scan: show whole body image and local or distant extent of disease. Activity on PET scan (measured by SUV activity) may predict response to chemotherapy and predict outcome (Figure 10)
- Bone scan: is used for detection of polyostotic disease and bone metastases (Figure 11)
Aspiration and trephine biopsy of bone marrow is mandatory to exclude bone marrow infiltration by morphologic analysis as well as molecular genetic detection of EWS/FLI1 (microscopic residual disease)
Biopsy: open surgical biopsy is the only way to confirm diagnosis of Ewing sarcoma. Biopsy samples are send to several labs:
- histopathology (morphology) pathologist see „small round blue cells“ pediatric poorly differentiated tumor with high proliferation activity (Ki67 index) (Figure 12)
- immunohistochemistry – patognomic is positivity of CD99, PAS positivity
- cytogenetic and molecular genetic studies (Figure 13)
For Ewing sarcoma is patognomic specific translocation inclusive EWS gene. It is positive in 90% of cases:
- EWS FLI1 t(11;22)(q24;q12)
- EWS FLI2 t(21;22)(q22;q12)
Based on initial work up patients are categorized into 3 groups:
- localized disease, with possible primary radical surgery
- locally advanced disease, primary radical surgery is not possible to perform
- metastatic disease
Differential diagnosis
- Inflammatory disease (chronic osteomyelitis) (Figure 14)
- Benign tumors (bone or soft tissue)
- Rheumatoid arthritis and autoaggressive diseases
- Other malignant bone tumors (osteosarcoma) (Figure 15)
- Hematologic malignancies (acute leukemia, lymphoma)
- Other malignant „small round blue cells“ pediatric tumors with bone metastases (neuroblastoma, rhabdomyosarcoma)
Therapy
Treatment of Ewing sarcoma needs multidisciplinary approach and all patients have to be treated in oncology centers specialized and experienced in diagnosis and therapy of musculoskeletal tumors.
The main goal of treatment is:
- to eliminate tumor mass
- to prevent recurrence
- to preserve function
The scheduling and integration of treatment modalities (chemotherapy, radiotherapy and surgery) must be carefully designed to provide a curative approach for the individual patient depends on stage and location of tumor (Figure 16)
A) Surgery
The role of surgery in management of Ewing sarcoma is diagnostic:
- initial open biopsy of primary tumor in case of relapse excision and verification of suspicious lesions
- therapeutic: local control of the tumor
Local control of Ewing sarcoma is crucial for prognosis and outcome of the patient.
Definitive surgical resection as a local therapy is preferred to radiotherapy and, if possible, is the first choice of local therapy. When tumor is localized on the extremities, limb salvage surgery is preferred. Indication for amputation is the same as for osteosarcoma except pathological fracture (Figure 17).
Curative surgery for bone tumors means bony margins of at least 1cm, with 2 to 5 cm margins recommended. Soft tissue margins at least 5mm in fat or muscle, with 2mm through fascial planes.
- timing is usually after 6 courses of neoadjuvant chemotherapy
- during definitive surgery the entire biopsy scar ca be excised en bloc with the tumor.
B) Chemotherapy
Effective systemic chemotherapy is necessary for cure of Ewing sarcoma. Induction (neoadjuvant or preoperative) chemotherapy is preferred over starting with local treatment (Figure 18).
Advantages of neoadjuvant chemotherapy:
- evaluation of effectiveness of chemotherapy ( % of necrosis)
- decreases volume of the tumor for local therapy
- chemotherapy eradicate possible micrometastases
- some bone healing occurs during chemo diminish the risk of pathological fracture
Postoperative adjuvant therapy is stratified according to:
- response to neoadjuvant chemotherapy
- evidence of metastatic disease
- radicality of surgery (radical – marginal – intralesional) (Figure 19)
C) Radiotherapy
Radiotherapy plays major role in obtaining local control of Ewing sarcoma due to its radiosensitivity. Radiotherapy can be used in combination with chemotherapy to achieve local control, but the radical surgery when feasible has to be regarded as the first choice of local control.
Indications for radiotherapy:
-
Radiotherapy before surgery:
- tumor progression on neoadjuvant chemo
- only marginal or intralesional surgery is possible
- risk of acute severe complications (spinal cord compression)
-
Postsurgical radiotherapy:
- intralesional or marginal surgery
- poor response to neoadjuvant chemotherapy
-
Definitive radiotherapy:
- inoperable tumors
- primary localisation on skull, face, vertebra, pelvis
- patient refusing surgery
- poor surgical risk
-
Palliative radiotherapy:
- pain control
- isolated bone metastases
- whole lung irradiation for lung metastases
D) Treatment of relapsed disease
...is usually difficult. Important factors for decision making regarding 2nd line therapy and prognosis are:
- time of relapse (early relapse – less than 6 months after the end of the treatment or late relapse)
-
localization of relapse:
- localized
- distant metastatic spread
- combined ( local and metastatic)
- type and intensity of the 1st line therapy
2nd line treatment options:
- surgery (if possible)
- 2nd line chemotherapy plus biphosphonates
- more intensive chemotherapy and high dose chemotherapy with autologous hematopoietic stem cell transplantation
- biology targeted therapy (TK inhibitors) as a part of clinical trials
- palliative metronomic chemotherapy
Prognosis and outcome
Predictive/prognostic factors (Figure 20)
- Tumor response to neoadjuvant chemotherapy (necrosis >90%)
- Evidence of metastatic disease
- Localization of metastases (lungs > bones >BM)
- Volume of primary tumor (>200 cm3) or size > 8 cm
- Axial localization of primary tumor (pelvis, spine, chest wall)
- Age (patients < 10 years have better outcome)
- Evidence of extraosseal soft tissue involvement (important for surgery)
- Radical tumor resection (margins R0 > R1 > R2)
5 years overall survival ES/PNET (Figure 21)
- localized disease 70%
- metastatic disease – lungs 30%
- metastatic disease (bones, bone marrow) < 10%
Author: Viera Bajčiová, MD, PhD
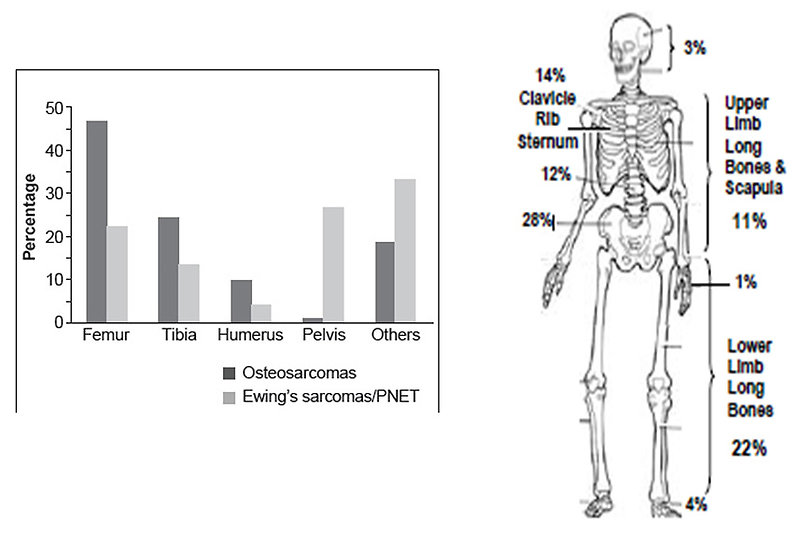 Figure 1: The most common location of Ewing sarcoma |
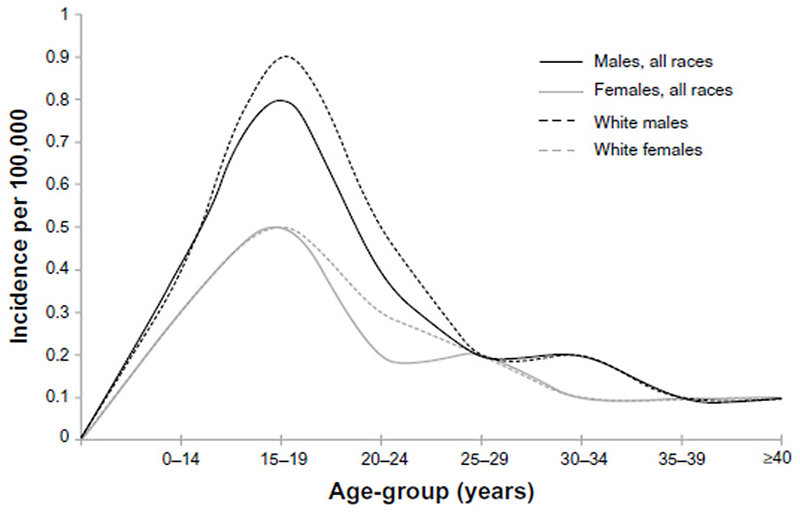 Figure 2: Incidence of Ewing sarcoma |
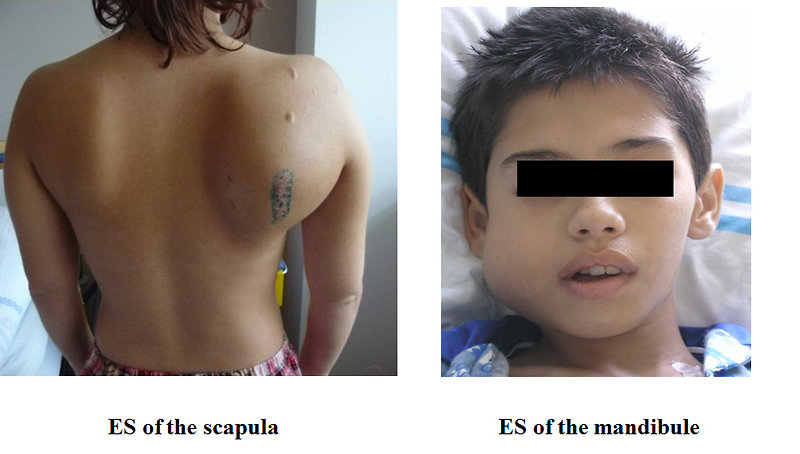 Figure 3: Clinical presentation of Ewing sarcoma |
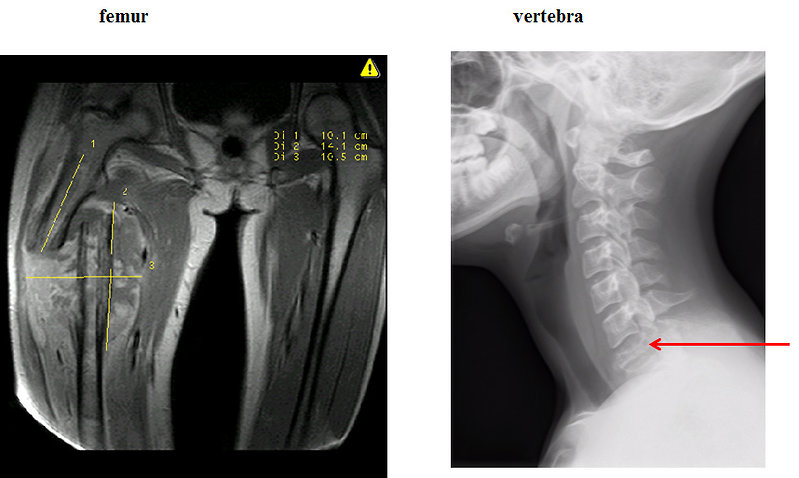 Figure 4: Pathologic fracture of Ewing sarcoma |
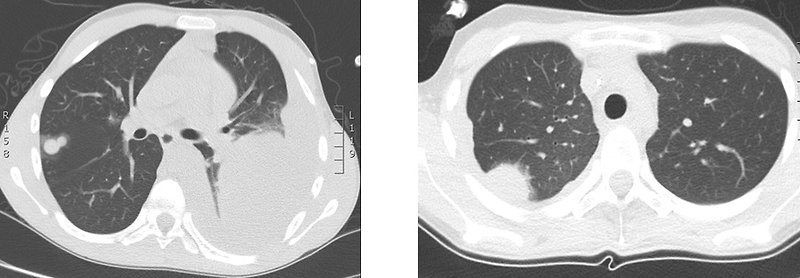 Figure 5. Lung metastases of Ewing sarcoma |
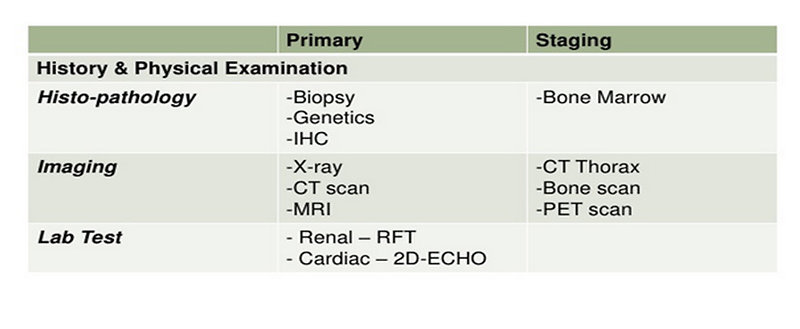 Figure 6: Diagnostic work up |
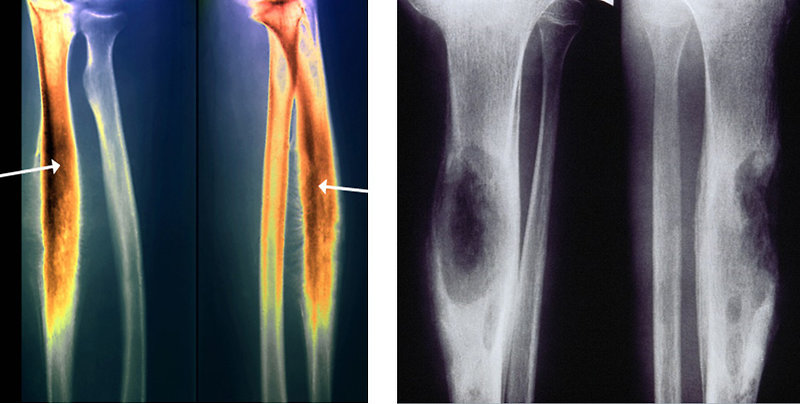 Figure 7: Plain X-ray |
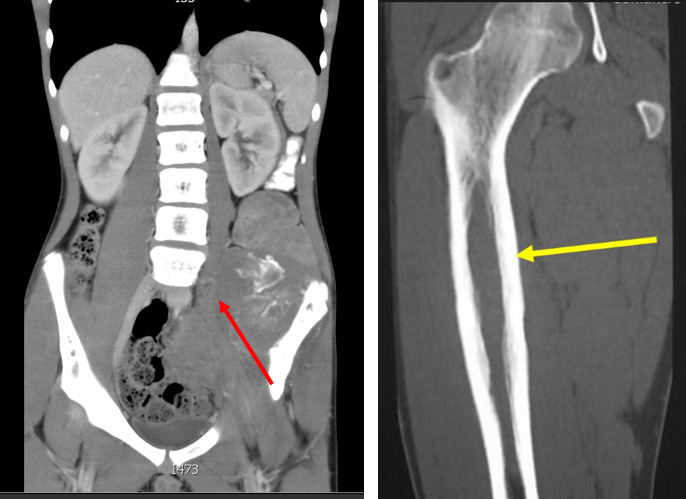 Figure 8: Ewing sarcoma CT scan |
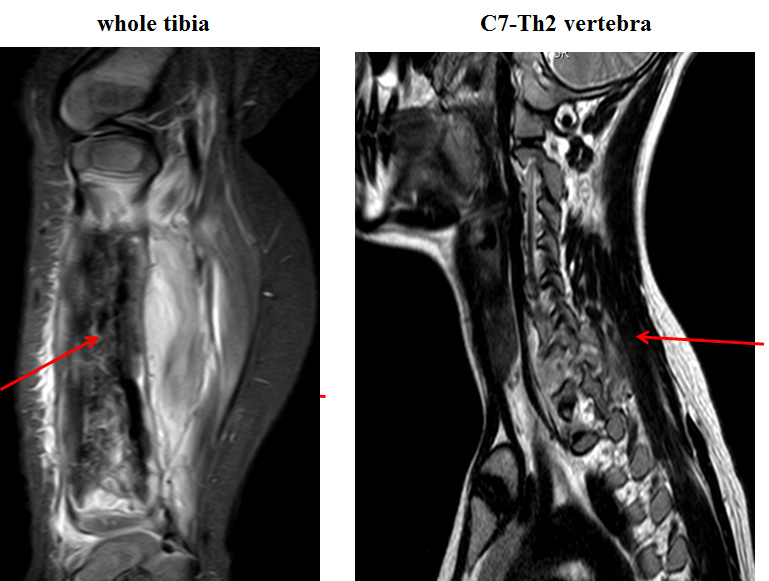 Figure 9: Ewing sarcoma MRI |
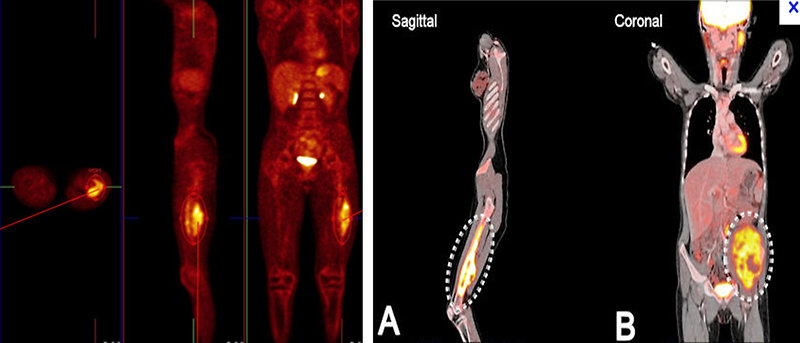 Figure 10 : Ewings sarcoma on 18 FDG PET scan |
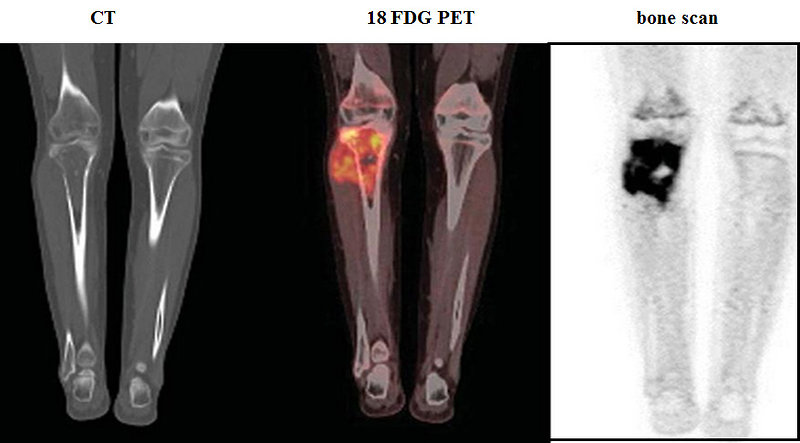 Figure 11: CT, PET and bones scan of Ewing sarcoma |
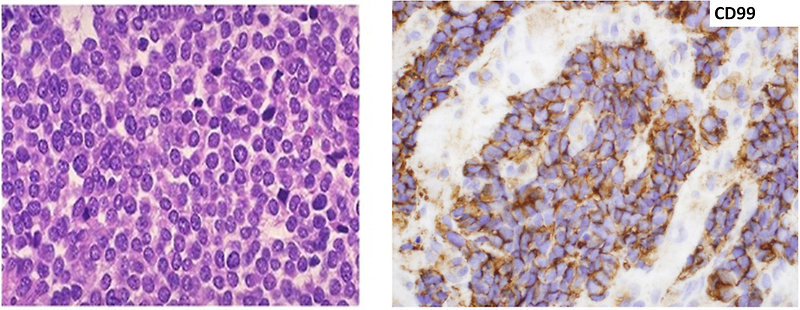 Figure 12 : Pathology – morphology of Ewing sarcoma |
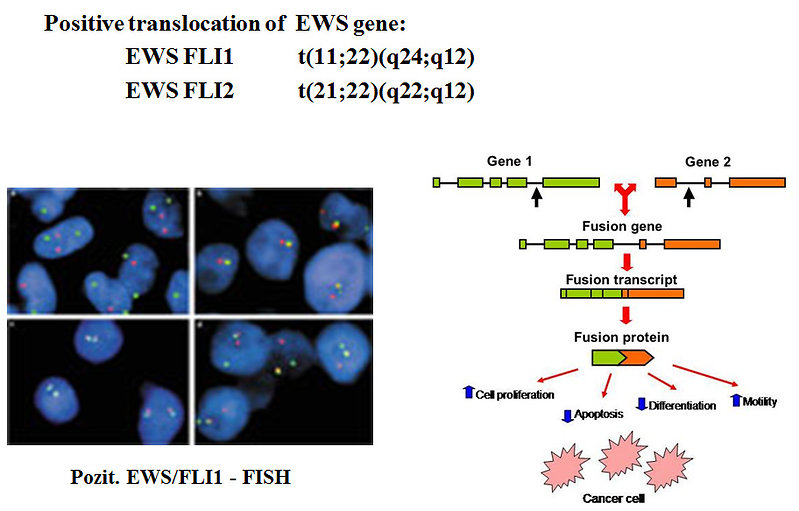 Figure 13: Typical ES/PNET translocation |
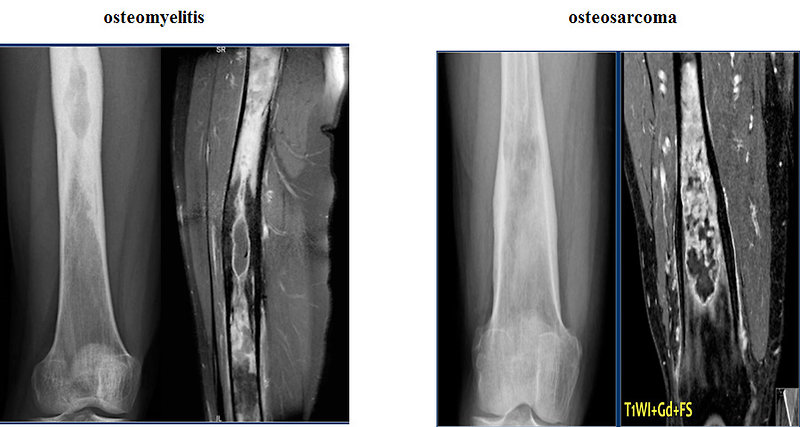 Figure 14: Differential diagnosis of bone lesions |
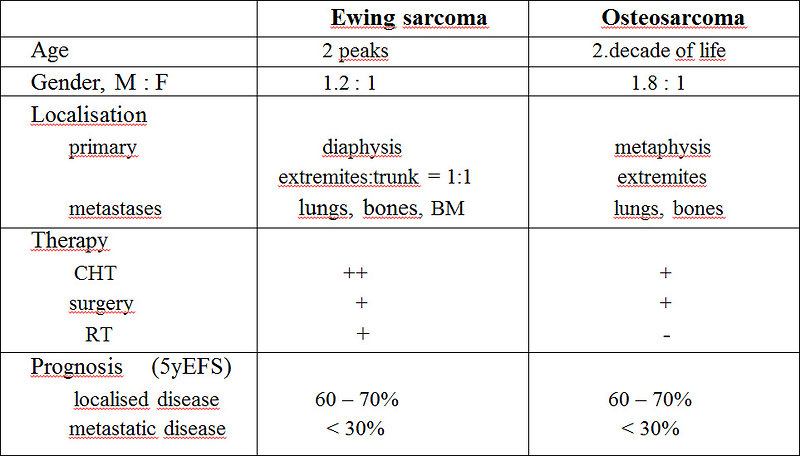 Figure 15: Differential diagnosis osteosarcoma vs Ewing sarcoma of the bone |
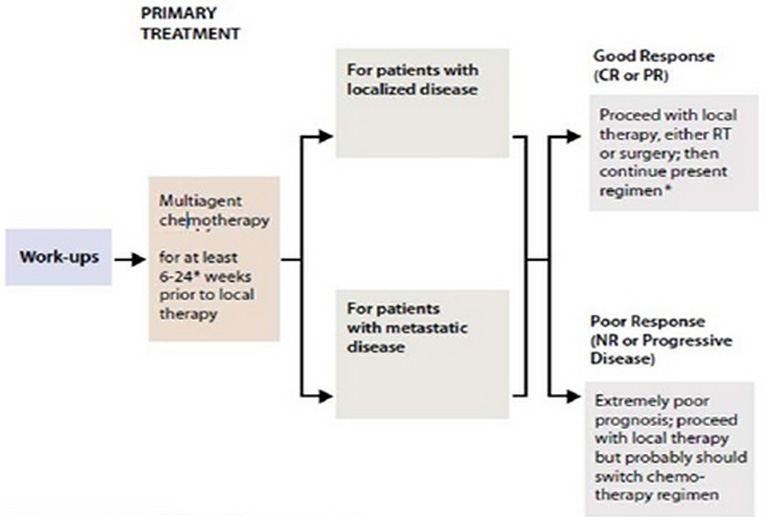 Figure 16: Overview of Ewing sarcoma treatment |
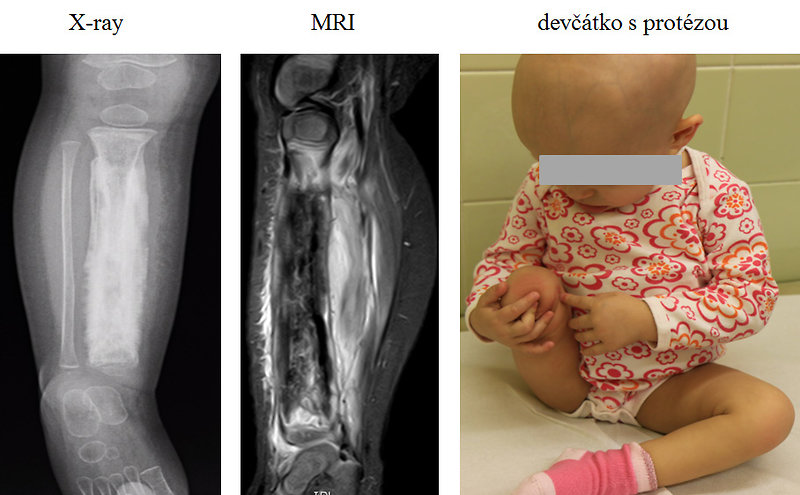 Figure 17: Ewing sarcoma - amputation |
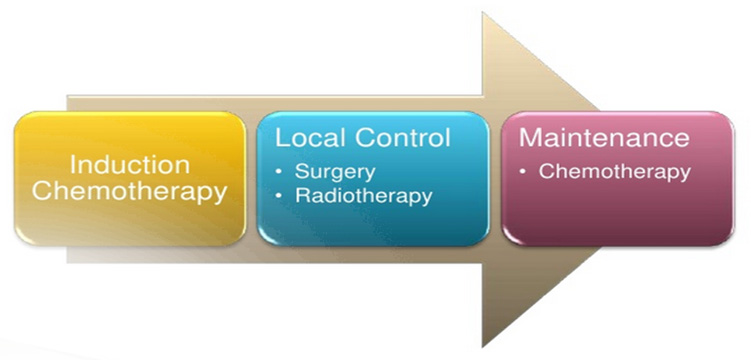 Figure 18: Treatment scheme |
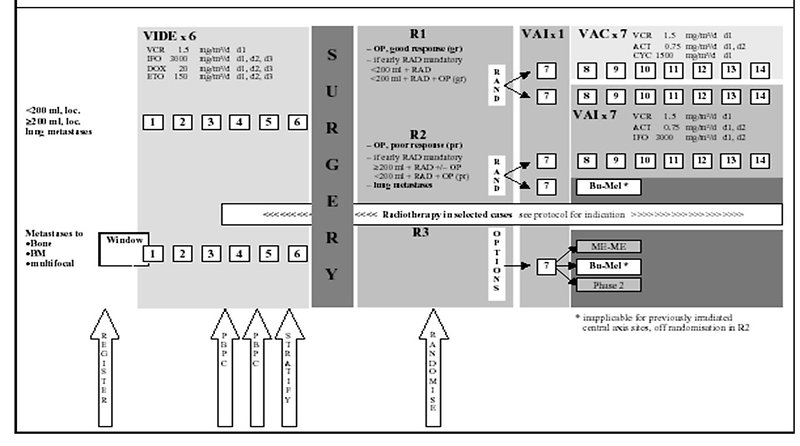 Figure 19: Treatment protocol for Ewing sarcoma/PNET |
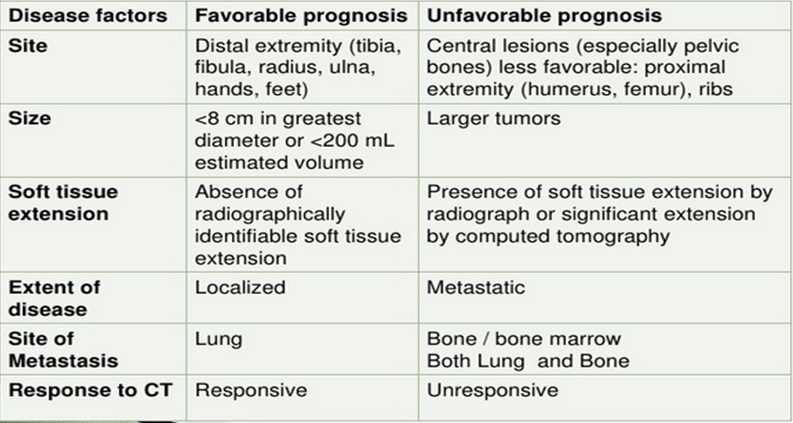 Figure 20: Prognostic factors |
 Figure 21: Survival of Ewing sarcoma |
|||